La distrofia corneale granulare (GCD) è una malattia corneale ereditaria caratterizzata da depositi granulari nello stroma corneale. Secondo la seconda classificazione internazionale delle distrofie corneali (IC3D 2015), è classificata come distrofia epitelio-stromale correlata a TGFBI2).
È causata da mutazioni puntiformi del gene TGFBI (cromosoma 5q31) e segue un modello di ereditarietà autosomica dominante. In base alla mutazione, si distinguono due tipi:
Classificazione
Mutazione principale
Altro nome / Vecchia denominazione
Deposito principale
GCD1
Arg555Trp (R555W)
Granulare classica / Groenouw tipo 1
Solo ialina
GCD2
Arg124His (R124H)
Distrofia corneale di Avellino
ialina + amiloide
La GCD2 è stata descritta nel 1988 come sottotipo distinto dalla GCD1, e poiché la prima famiglia proveniva dalla regione di Avellino in Italia, è stata chiamata distrofia corneale di Avellino 1). Successivamente, nel 1997, è stato identificato il gene causativo TGFBI e la mutazione R124H 3). Dalla seconda edizione della classificazione IC3D (2015), è stata ufficialmente denominata distrofia corneale granulare di tipo 2 (GCD2), con “Avellino” indicato come nome storico 2).
A livello internazionale, la distrofia corneale granulare è classificata nel gruppo delle distrofie epitelio-stromali. Questo gruppo comprende sei malattie correlate a TGFBI: granulare (GCD1 e GCD2), reticolare (LCD1 e LCD3A), Reis-Bücklers e Thiel-Behnke, tutte causate da diverse mutazioni puntiformi nel gene TGFBI sul cromosoma 5q31 2,4). Nella pratica clinica oftalmologica giapponese, queste sei malattie sono comunemente denominate collettivamente “distrofie corneali correlate a TGFBI”.
La distrofia corneale granulare fu descritta per la prima volta da Groenouw nel 1890, e all’epoca veniva semplicemente chiamata “tipo 1 di Groenouw”. Nel 1938 fu chiarita la distinzione dalla distrofia reticolare, e per lungo tempo è stata trattata come un’unica entità sotto il nome di “distrofia corneale granulare”. Nel 1988, una famiglia della regione di Avellino in Italia presentò una forma con caratteristiche sia granulari che reticolari, che in seguito fu separata come GCD2 (tipo Avellino) 1,2). Con la revisione della classificazione IC3D del 2015, è stato stabilito l’attuale sistema di classificazione e la nomenclatura basata sul genotipo è diventata lo standard internazionale 2).
Modalità di trasmissione: autosomica dominante. Mostra elevata penetranza
Tipo 1: comune in Europa e America. Raro in Giappone
Tipo 2: estremamente prevalente in Asia orientale, come Giappone e Corea. In Corea, la prevalenza è di circa 11,5 persone ogni 10.000 1)
Proporzione tra le distrofie corneali correlate a TGFBI: la GCD2 rappresenta il 72-91% in Corea e Giappone, il 36% negli Stati Uniti e il 3% in Polonia 1)
Dati di diagnosi genetica in Giappone: All’Università di Yamaguchi, in 21 anni dal 2000 al 2021, 234 pazienti con distrofia corneale sono stati diagnosticati geneticamente, e le quattro principali distrofie corneali (tipo granulare I e II, tipo reticolare I e IIIA, distrofia corneale gelatinosa a gocce, e tipo maculare) rappresentavano circa il 96% del totale. - Caratteristiche nell’Asia orientale: La distrofia corneale granulare è prevalentemente di tipo 2 (R124H) nell’Asia orientale. - Età di insorgenza: Gli eterozigoti per GCD2 presentano microopacità visibili solo alla lampada a fessura dall’età scolare, ma senza sintomi soggettivi. La diminuzione soggettiva dell’acuità visiva compare tipicamente tra i 40 e i 50 anni.
Differenza di sesso: È una malattia autosomica dominante, quindi non c’è differenza di sesso.
QCosa si intende per "granulare"?
A
Si riferisce a una condizione in cui si formano numerosi piccoli grumi bianchi o grigio-biancastri (depositi granulari) con confini netti nello stroma superficiale della parte centrale della cornea. Osservati direttamente con il microscopio a lampada a fessura, vengono descritti come a forma di briciole di pane, fiocchi di neve o confetti. I depositi derivano dalla proteina TGFBI mutata.
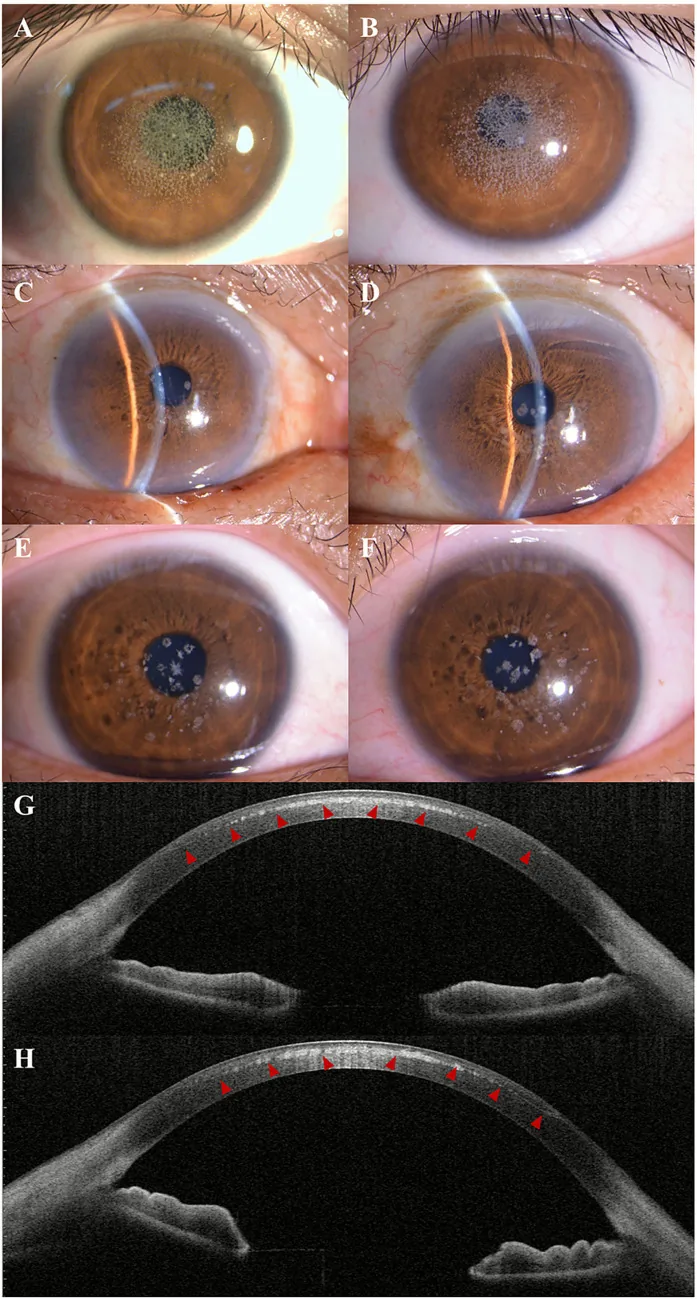
Kuang L, et al. Case Report: Post-LASIK exacerbation of granular corneal dystrophy type 2: a familial case with TGFBI mutation. Front Med (Lausanne). 2025. Figure 1. PMCID: PMC12722859. License: CC BY.
Nella fotografia con lampada a fessura, si osservano opacità granulari grigio-biancastre sparse e aggregate al centro e paracentro della cornea. L’AS-OCT mostra depositi iperriflettenti nello stroma corneale anteriore, indicando i reperti clinici della distrofia corneale granulare.
Asintomatico o lieve: Negli eterozigoti, nelle fasi iniziali e intermedie non si avverte una diminuzione dell’acuità visiva, e non è raro che venga scoperta casualmente durante controlli medici. Spesso la diminuzione della vista viene riferita solo dopo i 40-50 anni.
Abbagliamento e fotofobia: Quando le opacità coinvolgono l’area pupillare, si lamentano abbagliamento diurno e riduzione della sensibilità al contrasto.
Erosione corneale ricorrente: I depositi danneggiano la membrana di Bowman e la membrana basale epiteliale, causando dolore oculare acuto, lacrimazione e arrossamento durante il sonno o al risveglio.
Diminuzione dell’acuità visiva: Quando le aree trasparenti tra i depositi diventano opache, l’acuità visiva diminuisce progressivamente3).
Riduzione della sensibilità al contrasto: Spesso la sensibilità al contrasto diminuisce prima dell’acuità visiva (test dell’anello di Landolt).
Tendenza alla cecità diurna: Poiché la luce diffusa ha un forte impatto in ambienti luminosi, si lamenta abbagliamento all’aperto o durante la guida.
Difficile da correggere con occhiali o lenti a contatto: La dispersione dei depositi non migliora con la correzione refrattiva.
Negli omozigoti, si verifica una marcata riduzione della vista già in giovane età (4-7 anni) e il trattamento diventa necessario intorno ai 10 anni.
Reperti clinici (reperti osservati dal medico durante la visita)
Opacità granulari: Opacità granulari bianche o bianco-grigiastre, relativamente piccole e dai bordi netti, sparse nella parte centrale della cornea. Vengono descritte come a forma di briciole di pane o fiocchi di neve.
Profondità: Sottoepiteliali e nello strato superficiale dello stroma corneale. Non si estendono al limbo.
Sostanza depositata: Solo ialina. Si colora di rosso con la colorazione tricromica di Masson. Non contiene amiloide.
Progressione: Con l’età, il numero di granuli aumenta e i bordi diventano meno netti.
GCD2 (R124H)
Opacità granulari: Esordisce con opacità bianche o bianco-grigiastre più grandi di quelle della GCD1, dai bordi netti. Il fenotipo è vario: a forma di confetto, lineare, stellato, a clava, ecc.
Tipo misto: Talvolta si osservano anche sottili opacità lineari reticolari simili a quelle della distrofia corneale reticolare.
Sostanza depositata: Sia ialina che amiloide. Positivo alla colorazione tricromica di Masson e alla colorazione rosso Congo, e mostra birifrangenza giallo-verde al microscopio a luce polarizzata.
Progressione: Dopo i 25-30 anni, si aggiungono opacità bianche dense, a forma di bastoncino o stellata, nello strato medio dello stroma. Nello strato superficiale, i depositi diffusi a placca diventano più intensi, rendendo la PTK una buona indicazione3).
In entrambi i tipi, le opacità sono localizzate nella parte centrale della cornea e non si estendono alla periferia limbare. Di solito sono bilaterali e con poca differenza tra i due occhi.
Nelle mutazioni omozigoti, il fenotipo è marcatamente diverso.
Omozigoti GCD1: opacità reticolari bianche, quasi senza spazi, alla stessa profondità sottoepiteliale e nello stroma superficiale della cornea. Con il progredire, l’iride e la camera anteriore diventano inosservabili.
Omozigoti GCD2: opacità bianche rotonde e solide si formano su tutta la superficie corneale senza spazi, eccetto la periferia estrema. È così grave che il bianco è riconoscibile a occhio nudo, e solo la trasparenza del limbo è preservata3). I casi omozigoti sono distrofie corneali refrattarie che recidivano in un breve periodo di 1-2 anni anche dopo PTK e trapianto di cornea.
QIn che modo il decorso differisce tra omozigoti ed eterozigoti?
A
Gli omozigoti insorgono nella prima infanzia (4-7 anni) e progrediscono rapidamente. Opacità bianche si formano su tutta la superficie corneale senza spazi, e PTK o trapianto di cornea sono necessari intorno ai 10 anni. Anche dopo l’intervento, recidivano in 1-2 anni, con un decorso refrattario. Gli eterozigoti progrediscono lentamente e di solito mantengono una buona acuità visiva fino ai 40-50 anni.
La GCD è causata da mutazioni puntiformi nel gene TGFBI (cromosoma 5q31). Il gene TGFBI codifica per la proteina della matrice extracellulare TGFBIp (cheratoepitelina). La TGFBIp mutata ha una ridotta suscettibilità alla degradazione proteolitica e si accumula come depositi anomali insolubili nello stroma corneale1,5,7).
Il gruppo delle distrofie corneali associate a TGFBI include le seguenti2,4):
Distrofia corneale granulare tipo 1 (R555W)
Distrofia corneale granulare tipo 2 (R124H, ex Avellino)
Anamnesi familiare: Ereditarietà autosomica dominante; il 50% dei figli di un soggetto affetto ha rischio di sviluppare la malattia.
Omozigosi: Gli omozigoti per la stessa mutazione mostrano un fenotipo più grave.
Chirurgia corneale: La GCD2 può progredire rapidamente dopo un trauma corneale. L’opacità peggiora notevolmente dopo chirurgia refrattiva laser, in particolare dopo LASIK.1,8,9)
Etnia: La GCD2 è più comune in Asia orientale (Corea, Giappone). Si ipotizza un effetto fondatore genetico.3)
Fattori ambientali non chiari: L’esposizione ai raggi UV e il diabete non hanno un impatto diretto stabilito al momento.
La GCD è una malattia autosomica dominante ad alta penetranza. Se un caso indice viene diagnosticato, il 50% dei parenti di primo grado (genitori, fratelli, figli) può essere portatore della stessa mutazione. Identificare precocemente i portatori asintomatici in famiglia permette di evitare futuri interventi di chirurgia refrattiva e di pianificare visite di controllo regolari per monitorare la progressione.1,5) In particolare, nei giovani che desiderano sottoporsi a LASIK, si raccomanda vivamente un’attenta anamnesi familiare e, se necessario, un test genetico.
La diagnosi clinica si basa sull’osservazione di opacità granulari ben definite nello stroma anteriore alla lampada a fessura e su un’anamnesi familiare positiva. In presenza di opacità corneali bilaterali (depositi) senza iperemia o edema corneale, si sospetta una distrofia corneale.3)
Nella diagnosi differenziale delle distrofie corneali, si valuta innanzitutto se i depositi sono ‘ben definiti’ o ‘diffusi’.3) Se i depositi granulari sono ben definiti, si distingue tra GCD1 (piccoli) e GCD2 (grandi) in base alle dimensioni. Nella GCD2, la sclerosi diffusa mostra opacità diffuse a forma di nastro tra i depositi granulari; queste opacità diffuse sono una buona indicazione per PTK.3)
Lampada a fessura: Osservazione diretta di opacità granulari bianche ben definite. Si utilizzano anche la sclerosi diffusa, la retroilluminazione e la transilluminazione.
Microscopia confocale: si osservano opacità irregolari, iperriflettenti, simili a briciole di pane tra l’epitelio e lo strato di Bowman
Microscopia ultrasonica biomicroscopica (UBM): rileva granuli iperriflettenti nello stroma superficiale
Analisi della morfologia corneale: fornisce informazioni aggiuntive sulla densità delle opacità
Test genetico: l’analisi del gene TGFBI è utile per la diagnosi definitiva. In Giappone, dall’aprile 2020, è coperto dall’assicurazione sanitaria come test genetico per le distrofie corneali 3)
Fotografia del segmento anteriore: per il follow-up a lungo termine, è importante scattare fotografie di alta qualità del segmento anteriore alla prima visita e ad ogni controllo, conservandole nella cartella clinica
GCD1: depositi ialini che si colorano di rosso con la colorazione tricromica di Masson. Non contengono amiloide. Al microscopio elettronico, depositi a forma di bastoncino o trapezoidali
GCD2: si depositano sia ialina (positiva alla colorazione tricromica di Masson) che amiloide (positiva alla colorazione rosso Congo, giallo-verde al microscopio a luce polarizzata). Al microscopio elettronico si osservano depositi elettrondensi a bastoncino e fibrille amiloidi 1,7)
Distrofia corneale a reticolo tipo 1 (LCD1): mutazione TGFBI R124C. Opacità lineari e reticolari dovute a deposizione di amiloide nello stroma. Spesso associata a erosioni epiteliali ricorrenti3)
Distrofia corneale maculare (MCD): mutazione del gene CHST6. Ereditarietà autosomica recessiva. Opacità diffusa di tutta la cornea
Distrofia corneale di Reis-Bücklers: mutazione TGFBI R124L. Opacità a forma di mappa nello strato di Bowman
Distrofia corneale a macchie (FCD): mutazione PIP5K3. Piccole macchie bianche in tutto lo stroma, di solito asintomatiche
QIl test genetico è coperto dall'assicurazione?
A
Dall’aprile 2020, il test genetico per la distrofia corneale è coperto dall’assicurazione sanitaria. Tuttavia, è necessaria la certificazione della struttura, quindi le strutture in grado di eseguire il test sono limitate. Se i reperti clinici sono sospetti o si sta considerando un intervento di chirurgia refrattiva come LASIK, è consigliabile una diagnosi definitiva tramite test genetico.
Nelle fasi iniziali senza riduzione dell’acuità visiva o erosioni epiteliali ricorrenti, non è necessario alcun trattamento. Si considera un intervento chirurgico quando la riduzione della vista raggiunge l’area pupillare.
Lacrime artificiali: collirio a base di ialuronato di sodio allo 0,1% o 0,3% da usare 4-6 volte al giorno per ridurre secchezza e irritazione
Lenti a contatto morbide terapeutiche: per erosioni epiteliali ricorrenti, proteggono la superficie oculare e favoriscono la guarigione. Di solito vengono indossate tutto il giorno e richiedono una sostituzione regolare
Colliri antibiotici e unguenti oftalmici: per prevenire infezioni secondarie in caso di erosione epiteliale, usare levofloxacina 0,5% collirio 3-4 volte al giorno e unguento oftalmico di ofloxacina prima di dormire
Soluzione salina ipertonica (collirio o unguento oftalmico al 5% di cloruro di sodio): può essere utilizzata come trattamento coadiuvante per ridurre l’edema epiteliale
Omozigoti per GCD2: recidiva circa 18 mesi dopo la prima PTK, e dopo la seconda, terza e successive entro circa 3 mesi1)
Eterozigoti per GCD2: la recidiva dopo PTK è relativamente lenta, con una media di 38,4 mesi1)
Uso concomitante di mitomicina C (MMC): l’uso di MMC durante la PTK non è raccomandato. La MMC induce l’apoptosi dei cheratociti dello stroma corneale, riducendo le cellule responsabili del riassorbimento e della degradazione di TGFBIp, con possibile accelerazione della recidiva1)
Casi di peggioramento dopo LASIK: la PTK è possibile, ma l’effetto è maggiore dopo la rimozione del flap LASIK1,8)
Gestione post-operatoria: dopo PTK, utilizzare colliri antibiotici (levofloxacina 0,5%) e corticosteroidi (fluorometolone 0,1%) 4 volte al giorno fino alla guarigione epiteliale, quindi ridurre gradualmente. La guarigione epiteliale richiede solitamente 3–5 giorni.
DALK (cheratoplastica lamellare profonda) nella pratica
La DALK è una tecnica che preserva l’endotelio: utilizzando la tecnica della grande bolla (big bubble technique), si asporta lo stroma fino alla membrana di Descemet e si sutura lo stroma del donatore. Poiché non vi è rischio di rigetto endoteliale, la prognosi a lungo termine è considerata migliore rispetto alla PK3). Secondo uno studio di Kitazawa et al., la DALK per le distrofie corneali associate a TGFBI (incluse le forme granulare e reticolare) mostra una buona acuità visiva a 5 anni e un alto tasso di sopravvivenza dell’innesto10). In Giappone, è eseguibile come trattamento coperto dall’assicurazione sanitaria.
La GCD è una controindicazione per LASIK, LASEK, PRK e SMILE. Dopo l’intervento, l’opacità corneale può peggiorare rapidamente, causando una grave riduzione della vista 1,8,9). Dopo LASIK, si formano numerosi piccoli depositi granulari tra il flap e lo stroma. Il peggioramento è più grave con LASIK rispetto a PRK e l’acuità visiva finale è peggiore 1,8). Rapporti di casi dalla Corea e dal Giappone descrivono molti pazienti asintomatici prima dell’intervento che hanno sviluppato una marcata opacità corneale da alcuni mesi ad anni dopo LASIK, richiedendo PTK o trapianto di cornea8,9).
QCosa succede se la GCD viene diagnosticata dopo aver subito LASIK?
A
Nei casi di GCD insorta dopo LASIK, si formano rapidamente depositi granulari tra il flap e lo stroma. Le opzioni terapeutiche includono PTK dopo rimozione del flap LASIK, DALK e PK, da considerare in quest’ordine. È importante consultare tempestivamente un oculista specialista.
Il gene TGFBI codifica per la proteina della matrice extracellulare TGFBIp (cheratoepitelina, 68 kDa). La TGFBIp è coinvolta nell’adesione, migrazione e proliferazione cellulare ed è espressa anche nello stroma corneale normale 1,5,7). Quando il gene TGFBI presenta una mutazione, la TGFBIp mutata ha una ridotta suscettibilità alla degradazione proteolitica e si accumula come depositi insolubili nello stroma corneale5,7).
La GCD1 è causata dalla mutazione Arg555Trp (R555W). La TGFBIp mutata si deposita come ialina nello stroma corneale superficiale. Non contiene amiloide 3).
La GCD2 è quasi esclusivamente causata dalla mutazione Arg124His (R124H) 1,5). Nella GCD2 si depositano sia ialina che amiloide.
Alterazione dell’autofagia: Nella GCD2 è stata riportata un’alterazione dell’autofagia, che riduce la degradazione della TGFBIp e ne favorisce l’accumulo 1,5)
Disfunzione mitocondriale: È stato suggerito che la TGFBIp mutata stessa possa influenzare i fibroblasti corneali e causare disfunzione mitocondriale 1)
Effetto della neovascolarizzazione corneale: Nelle aree con neovascolarizzazione corneale, i depositi tendono a ridursi e a essere riassorbiti. Questa osservazione supporta il meccanismo per cui i depositi si concentrano nella parte centrale della cornea, priva di apporto vascolare1)
Dopo LASIK, la TGFBIp si deposita rapidamente tra il flap e il letto stromale. Si ritiene che ciò sia dovuto al fatto che la manipolazione chirurgica nella parte centrale della cornea favorisce l’accumulo di TGFBIp mutata1,8). Poiché l’incisione corneale durante la chirurgia della cataratta (vicino al limbo) non provoca un aggravamento, si ipotizza che la distanza dal limbo vascolarizzato sia correlata1). Le osservazioni patologiche di Awwad et al. suggeriscono che i depositi formati dopo LASIK si accumulano insieme all’attivazione dei cheratociti associata alla reazione di guarigione della ferita all’interfaccia tra flap e letto stromale8).
I depositi di GCD1 appaiono al microscopio ottico come sostanze eosinofile omogenee e si colorano di rosso con la colorazione tricromica di Masson. A livello di microscopia elettronica, si riconoscono come strutture ad alta densità elettronica a forma di bastoncino o trapezoidale, con diametro di 100-500 nm6).
Nella GCD2, oltre ai depositi ialini, si osservano fibre amiloidi (diametro 8-10 nm). Le fibre amiloidi si colorano di rosso-arancio con la colorazione rosso Congo e mostrano birifrangenza verde mela al microscopio a luce polarizzata6). La doppia caratteristica di colorazione è utile per la diagnosi patologica definitiva di GCD2.
Secondo l’analisi proteomica di Poulsen et al., nella cornea dei pazienti con GCD2, la TGFBIp mutata R124H è meno suscettibile al taglio da parte degli enzimi proteolitici rispetto alla TGFBIp normale, e specifici frammenti C-terminali si accumulano selettivamente6). Si suggerisce che questa resistenza al taglio possa essere alla base della formazione di fibre ialine e amiloidi.
È stato riportato che il cloruro di litio riduce la produzione della proteina TGFBI. La terapia combinata con melatonina e rapamicina inibisce l’espressione della proteina TGFBI e allo stesso tempo attiva l’autofagia, promuovendo potenzialmente la degradazione della TGFBIp mutata1,5).
Il silenziamento dell’espressione di TGFBI mutata mediante small interfering RNA (siRNA) o short hairpin siRNA (shRNA) è in fase di studio preclinico. Anche la tecnologia di editing genomico CRISPR/Cas9 è un candidato, ma l’effetto off-target indesiderato sugli alleli normali o su altri geni rimane una sfida1,5).
Elettrolisi corneale: è stato riportato l’uso sperimentale per i casi di recidiva dopo trapianto di cornea. I risultati a lungo termine sono sconosciuti.
Supporto diagnostico tramite machine learning: è stato riportato lo sviluppo di un modello di IA per identificare automaticamente la GCD dalle foto del segmento anteriore.
Terapia con chaperoni: sono in corso studi di base su chaperoni chimici (come l’acido 4-fenilbutirrico) che aiutano il corretto ripiegamento del TGFBIp mutato3).
Foglietti epiteliali corneali derivati da cellule iPS: il trapianto di foglietti epiteliali corneali prodotti da cellule iPS del paziente è studiato come opzione futura.
A livello nazionale, la Società Giapponese di Oftalmologia e la Società Giapponese della Cornea stanno discutendo la creazione di un registro nazionale per le distrofie corneali correlate a TGFBI. Con la copertura assicurativa dei test genetici dal 2020, il numero di casi diagnosticati geneticamente è aumentato e si stanno accumulando dati di follow-up a lungo termine sui modelli di mutazione e fenotipi specifici dei giapponesi3,11). In futuro, ci si aspetta lo sviluppo di un sistema clinico basato sull’evidenza, dallo screening dei portatori alla valutazione dell’idoneità per la chirurgia refrattiva.
Per i pazienti con GCD, le seguenti indicazioni sullo stile di vita sono importanti.
Protezione dai raggi UV: utilizzare occhiali da sole o occhiali con protezione UV quando si esce per ridurre l’esposizione della cornea ai raggi UV.
Evitare traumi oculari: contusioni o corpi estranei possono indurre erosioni epiteliali ricorrenti. Utilizzare occhiali protettivi durante sport o lavori.
Visite regolari: anche gli eterozigoti dovrebbero sottoporsi a esame con lampada a fessura e test dell’acuità visiva una volta all’anno.
Esame dei familiari: raccomandare lo screening familiare anche ai parenti di primo grado.
Lenti a contatto: le lenti morbide sono generalmente possibili, ma ridurre il tempo di utilizzo e rispettare rigorosamente la sostituzione periodica.
Evitare la chirurgia refrattiva: gli interventi di tipo LASIK sono assolutamente controindicati. Scegliere la correzione con occhiali o lenti a contatto.
Chang MS, Jun I, Kim EK. Mini-Review: Clinical Features and Management of Granular Corneal Dystrophy Type 2. Korean journal of ophthalmology : KJO. 2023;37(4):340-347. doi:10.3341/kjo.2023.0032. PMID:37336511; PMCID:PMC10427907.
Weiss JS, Møller HU, Aldave AJ, et al. IC3D classification of corneal dystrophies-Edition 2. Cornea. 2015;34(2):117-159. doi:10.1097/ICO.0000000000000307. PMID:25564336.
Lisch W, Weiss JS. Clinical and genetic update of corneal dystrophies. Experimental eye research. 2019;186:107715. doi:10.1016/j.exer.2019.107715. PMID:31301286.
Han KE, Choi SI, Kim TI, Maeng YS, Stulting RD, Ji YW, et al. Pathogenesis and treatments of TGFBI corneal dystrophies. Progress in retinal and eye research. 2016;50:67-88. doi:10.1016/j.preteyeres.2015.11.002. PMID:26612778.
American Academy of Ophthalmology Cornea/External Disease Panel. Corneal Edema and Opacification Preferred Practice Pattern®. Ophthalmology. 2024;131(4):P1-P79.
Poulsen ET, Dyrlund TF, Runager K, et al. Proteomics of granular corneal dystrophy type 2 reveals altered TGFBIp proteolytic processing. J Proteome Res. 2014;13(10):4659-4667.
Dinh R, Rapuano CJ, Cohen EJ, Laibson PR. Recurrence of corneal dystrophy after excimer laser phototherapeutic keratectomy. Ophthalmology. 1999;106(8):1490-1497.
Awwad ST, Di Pascuale MA, Hogan RN, Forstot SL, McCulley JP, Cavanagh HD. Avellino corneal dystrophy worsening after laser in situ keratomileusis: further clinicopathologic observations and proposed pathogenesis. American journal of ophthalmology. 2008;145(4):656-61. doi:10.1016/j.ajo.2007.12.008. PMID:18243154; PMCID:PMC2661203.
Jun RM, Tchah H, Kim TI, et al. Avellino corneal dystrophy after LASIK. Ophthalmology. 2004;111(3):463-468.
Kitazawa K, Kawasaki S, Shinomiya K, et al. Safety of anterior lamellar keratoplasty for recurrent TGFBI corneal dystrophy. Br J Ophthalmol. 2016;100(4):448-454.