La distrofia corneale a gocce gelatinose (gelatinous drop-like corneal dystrophy: GDLD) è una malattia corneale ereditaria caratterizzata da depositi di amiloide sotto l’epitelio corneale, che porta a una marcata riduzione bilaterale dell’acuità visiva.
Questa malattia è stata riportata per la prima volta nel 1914 da Nakaizumi, e dal 1932, quando Kiyosawa la chiamò «degenerazione corneale a gocce gelatinose», è conosciuta con questo nome. Nella classificazione IC3D (International Committee for Classification of Corneal Dystrophies) è classificata come distrofia epiteliale, con l’abbreviazione GDLD.
Il gene responsabile, TACSTD2 (tumor-associated calcium signal transducer 2), è stato identificato nel 1999 da Tsujikawa e collaboratori. È un gene a singolo esone situato sul cromosoma 1p324).
Prevalenza: Rara a livello mondiale, ma segnalata come relativamente comune in Giappone 1). Si ritiene che l’incidenza sia diminuita a causa della riduzione dei matrimoni tra consanguinei 2)
Differenze regionali: Relativamente comune in Giappone, quasi mai segnalata in Europa e Nord America
Mutazione Q118X: Mutazione fondatrice nei pazienti giapponesi, che rappresenta oltre l’80% dei cromosomi patogeni 2)
Età di insorgenza: Spesso si manifesta entro i 20 anni
Nel 2019 è stata riconosciuta come malattia rara specifica “Distrofia corneale a gocce gelatinose” ed è diventata idonea per l’assistenza finanziaria alle spese mediche 2). Nell’ambito del progetto di ricerca sulle politiche per le malattie refrattarie del Ministero della Salute, del Lavoro e del Welfare, sono stati sviluppati criteri diagnostici e una classificazione della gravità 2).
QLa GDLD si manifesta anche al di fuori del Giappone?
A
La GDLD è stata segnalata in tutto il mondo, ma è più comune in Giappone. In Europa e Nord America non ci sono quasi casi. Sono state segnalate oltre 20 mutazioni del gene TACSTD2, che mostrano eterogeneità genetica. In Giappone, la mutazione Q118X situata su 1p32 è frequente come mutazione fondatrice e rappresenta oltre l’80% dei cromosomi patogeni nei pazienti giapponesi.
Spesso si manifesta entro i 20 anni. I seguenti sintomi compaiono fin dall’infanzia.
Fotofobia: Sintomo marcato fin dall’inizio
Sensazione di corpo estraneo: A causa delle elevazioni gelatinose sulla superficie corneale
Lacrimazione: Associata a sintomi di irritazione
Riduzione della vista: Peggiora gradualmente con la progressione della deposizione di amiloide. Diventa marcata dopo l’età adulta
Con l’età, il numero e le dimensioni dei depositi di amiloide aumentano. Diventano depositi grigio-biancastri o gialli e alla fine ricoprono la maggior parte della cornea, in particolare nell’area della rima palpebrale 2). Si verificano invasione vascolare dalla periferia, marcata riduzione dell’acuità visiva e dolore oculare, e si aggiungono problemi estetici, riducendo notevolmente la qualità di vita del paziente.
Reperti clinici (reperti confermati dal medico durante la visita)
L’opacità corneale è classificata in quattro tipi in base alla morfologia. Questi possono essere distinti mediante esame del segmento anteriore con microscopio a lampada a fessura2,3).
Tipo gelso
typical mulberry type: Il tipo più tipico.
Parte centrale della cornea: Lesioni rilevate grigio-biancastre si aggregano, somigliando a un gelso.
Amiloide subepiteliale: Elevazioni gelatinose bianco-lattee e traslucide aumentano dal centro verso la periferia.
Tipo cheratopatia a banda
band-keratopathy type: Può essere osservato nelle fasi iniziali.
Fessura interpalpebrale: Opacità superficiale. Presenta reperti simili alla cheratopatia a banda.
Lesioni congiuntivali: Possono essere presenti anche lesioni della congiuntiva.
Tipo kumquat
kumquat-like type: Frequente nei casi avanzati.
Depositi giallo-biancastri diffusi: L’intera cornea diventa gialla, somigliando a un kumquat.
Invasione vascolare: Può essere accompagnata da neovascolarizzazione superficiale.
Tipo di opacità stromale
tipo di opacità stromale : stadio più avanzato.
Estensione allo stroma : la lesione si estende allo stroma corneale.
Invasione vascolare : le elevazioni gelatinose bianco-giallastre sono accompagnate da invasione vascolare.
Ide et al. hanno riportato lo spettro clinico dettagliato di 34 casi giapponesi e hanno mostrato che, nonostante la stessa mutazione del gene TACSTD2 (omozigote Q118X), coesistono quattro fenotipi3).
Inoltre, sono presenti i seguenti reperti caratteristici:
Colorazione ritardata alla fluoresceina (delayed staining) : nonostante l’assenza di danno epiteliale corneale, a causa dell’aumentata permeabilità dovuta a una formazione incompleta delle giunzioni strette, si osserva fluorescenza pochi minuti dopo l’instillazione di fluoresceina2)
Assottigliamento epiteliale : l’epitelio corneale è assottigliato nelle aree delle elevazioni gelatinose
Invasione vascolare : si osserva invasione vascolare superficiale nella regione periferica della cornea
A causa dell’anomalia del gene TACSTD2, la normale localizzazione intracellulare della Claudina-1 e della Claudina-7, proteine strutturali delle giunzioni strette nell’epitelio corneale, viene persa e la funzione di barriera epiteliale si riduce5). Di conseguenza, proteine come la lattoferrina dalle lacrime penetrano nella cornea, formano fibrille amiloidi e si depositano sotto l’epitelio. Nakatsuka et al. hanno dimostrato mediante analisi di biologia molecolare di famiglie giapponesi che TACSTD2 è essenziale per la normale localizzazione delle claudine e hanno chiarito che la patologia della GDLD è dovuta a una disfunzione delle giunzioni strette5).
Anamnesi familiare : a causa dell’ereditarietà autosomica recessiva, esiste il rischio di sviluppare la malattia se entrambi i genitori sono portatori.
Discendenza giapponese: la mutazione nonsenso Q118X è una mutazione fondatrice in Giappone, rappresentando oltre l’80% dei cromosomi patogeni2)
Matrimonio tra consanguinei: di solito i genitori del probando sono consanguinei. Tuttavia, la malattia può manifestarsi anche in eterozigoti composti nati da matrimoni tra famiglie non imparentate.
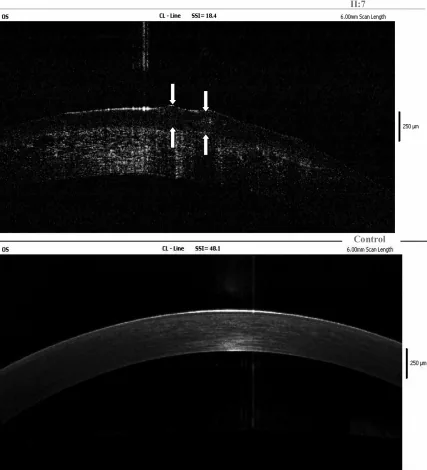
Yang Jing, Chun Liu, Liya Wang A novel TACSTD2 mutation identified in two Chinese brothers with gelatinous drop-like corneal dystrophy 2009 Aug 14 Mol Vis. 2009 Aug 14; 15:1580-1588 Figure 5. PMCID: PMC2728569. License: CC BY.
L’esame OCT nel dominio di Fourier della cornea sinistra del probando mostra depositi di amiloide nell’epitelio corneale e nello stroma superficiale; le frecce indicano la posizione delle lesioni a goccia gelatinosa. Ciò corrisponde ai depositi di amiloide trattati nella sezione «4. Diagnosi e metodi di esame».
Criteri diagnostici (gruppo di studio del Ministero della Salute)
I criteri diagnostici per la GDLD sono stati stabiliti dal gruppo di ricerca del progetto di politica sanitaria per le malattie refrattarie del Ministero della Salute, del Lavoro e delle Politiche Sociali, intitolato «Creazione e diffusione di linee guida cliniche per le malattie rare del segmento anteriore»2). Una diagnosi «Definite» secondo questi criteri rende la malattia eleggibile come malattia rara designata.
A. Sintomi (almeno uno dei seguenti)
Riduzione dell’acuità visiva
Fotofobia
Sensazione di corpo estraneo
Lacrimazione
B. Reperti degli esami
Si osservano accumuli di depositi amiloidi grigio-biancastri rilevati (a forma di mora) al di sotto dell’epitelio corneale nella parte centrale della cornea e della rima palpebrale di entrambi gli occhi.
Si osserva una colorazione ritardata (delayed staining): la fluorescenza appare alcuni minuti dopo la colorazione con fluoresceina, nonostante l’assenza di danno epiteliale corneale.
Si osserva un’invasione vascolare superficiale nella regione periferica della cornea.
C. Diagnosi differenziale : escludere l’amiloidosi corneale secondaria e la cheratopatia climatica a gocce.
Le condizioni per la categoria Certo sono soddisfatte se è presente uno dei seguenti 2):
Casi che soddisfano il criterio D, presentano uno dei criteri A, soddisfano il criterio B1 e consentono di escludere le malattie da differenziare in C.
Casi che soddisfano il criterio D, presentano uno dei criteri A, soddisfano il criterio B2 o B3, soddisfano il criterio E e consentono di escludere le malattie da differenziare in C.
B1 (depositi a forma di mora) è un reperto molto caratteristico; nei casi tipici la diagnosi non è difficile. Nei casi atipici, la diagnosi viene posta combinando i criteri A–C con l’esame genetico (E) 2).
Classificazione della gravità (secondo la notifica delle malattie rare designate)
La gravità viene classificata in gradi da I a IV in base alla migliore acuità visiva corretta dell’occhio migliore 2).
Gravità
Criterio
Assistenza medica
Grado I
Coinvolgimento di un solo occhio, l’altro occhio sano
×
Grado II
Coinvolgimento di entrambi gli occhi, migliore acuità visiva corretta ≥ 0,3
×
Grado III
Coinvolgimento di entrambi gli occhi, migliore acuità visiva corretta ≥ 0,1 e < 0,3
○
Grado IV
Coinvolgimento di entrambi gli occhi, migliore acuità visiva corretta < 0,1
○
Una diagnosi di Definite rende la malattia eleggibile come malattia rara designata, e per i gradi di gravità III e superiori è possibile ricevere un sussidio per le spese mediche 2). Se a causa di glaucoma secondario ecc. si verifica un restringimento del campo visivo (campo visivo centrale residuo ≤ 20 gradi con bersaglio Goldmann I/4) nell’occhio migliore, la gravità viene aumentata di un grado.
Esame con lampada a fessura: Osservare le lesioni rilevate grigio-biancastre dal centro della cornea alla regione della rima palpebrale, distinguendo i 4 tipi morfologici (a mora, a banda, a kumquat, opacità stromale).
Test di permeabilità alla fluoresceina (colorazione ritardata) : a causa della disfunzione delle giunzioni strette, il colorante penetra rapidamente nel tessuto corneale 2)
Test genetico TACSTD2 : dall’anno fiscale 2020 è coperto dall’assicurazione come test genetico per le distrofie corneali (D006-20) 2). TACSTD2 è un gene a singolo esone, facile da ricercare. Particolarmente utile per la diagnosi di casi atipici
Esame istologico (sezione corneale) : la colorazione con rosso Congo produce un colore rosso-arancio e, al microscopio a polarizzazione, mostra una birifrangenza verde mela, confermando l’amiloide
Amiloidosi corneale secondaria : deposito di amiloide dovuto a irritazione cronica come trichiasi, entropion, protrusione del cheratocono, uso di lenti a contatto rigide. L’assenza di storia familiare e la presenza di infiammazione cronica della superficie oculare sono punti di differenziazione. Può presentarsi con rilievi gelatinosi o aspetto reticolare; per la conferma è necessario l’esame istologico
Cheratopatia climatica a gocce : comune negli uomini di età superiore ai 40 anni. Osservata in regioni desertiche o estremamente fredde, causata da UV e secchezza. Presenta lesioni corneali rilevate di colore giallo o grigio-bianco
Degenerazione corneale a bandelletta : deposito di sali di calcio sotto l’epitelio. Inizia alla periferia alle ore 3 e 9 e progredisce verso il centro
Distrofia corneale reticolare di tipo I : mutazione R124C del gene TGFBI, eredità autosomica dominante. Presenta opacità fibrose ramificate nello stroma corneale
Distrofia corneale maculare : anomalia del gene CHST6, eredità autosomica recessiva. Opacità diffusa a vetro smerigliato
QIl test genetico è coperto dall'assicurazione?
A
Il test genetico TACSTD2 è coperto dall’assicurazione dal 2020 come «Test genetico per le distrofie corneali (D006-20)». Tuttavia, la struttura deve ottenere la certificazione dopo aver predisposto un sistema per eseguire il test al suo interno. Poiché TACSTD2 è un gene a singolo esone, facile da ricercare, e oltre l’80% dei pazienti giapponesi presenta la mutazione fondatrice Q118X, questo test è particolarmente utile per la diagnosi di casi atipici 2).
Il trattamento della GDLD viene scelto in base all’estensione dell’opacità e al grado di deficit visivo. A causa della natura ereditaria della malattia, il tasso di recidiva è estremamente elevato con qualsiasi terapia, rappresentando la sfida più grande 2). Non sono rari i casi di cecità dovuti a complicanze di multipli trapianti di cornea o glaucoma secondario.
Lacrime artificiali : utilizzate come terapia sintomatica per alleviare i sintomi di irritazione superficiale.
Uso continuo di lenti a contatto morbide terapeutiche (SCL) : può sopprimere la recidiva delle lesioni gelatinose e prolungare l’intervallo tra gli interventi chirurgici.
L’uso continuo di SCL terapeutiche è considerato un trattamento conservativo e adiuvante 6). Nel 2020, Maeno et al. hanno dimostrato in uno studio prospettico osservazionale su pazienti con GDLD che l’uso di SCL terapeutiche sopprime significativamente la recidiva delle lesioni gelatinose bianco-grigiastre o gialle 7). È raccomandato anche per la prevenzione delle recidive postoperatorie.
Cheratectomia superficiale terapeutica con laser ad eccimeri : trattamento di prima linea per le opacità superficiali.
Indicazioni : Proliferazioni gelatinose superficiali lievi o moderate. Combinato con raschiamento manuale. Risultati a lungo termine 8,9).
Trapianto di cornea
Superficiale, cheratoplastica lamellare profonda (DALK), perforante (PKP) : indicate nei casi avanzati.
Tasso di recidiva : Dopo cheratoplastica perforante, la recidiva è alta (97% entro 4 anni). La DALK ha il vantaggio di preservare l’endotelio.
Trapianto limbare
Trapianto di cellule staminali limbari, cheratoplastica epiteliale : in combinazione con trapianto di cornea.
Scopo : Ricoprire la superficie oculare con l’epitelio corneale del trapianto e prevenire la reinvasione dell’epitelio dell’ospite 10,11).
Diversi rapporti dal Giappone hanno documentato i risultati a lungo termine della PTK. Ōura et al. hanno mostrato i risultati a lungo termine della PTK in pazienti con GDLD, riportando l’utilità nel prolungare l’intervallo prima della recidiva 8). Hieda et al. hanno analizzato in dettaglio i tempi di recidiva e gli esiti clinici dopo PTK in uno studio multicentrico giapponese 9).
La combinazione con il trapianto di cellule staminali limbari (LSCT) è un approccio di origine giapponese riconosciuto a livello mondiale. Nel 2002, Shimazaki et al. hanno riportato l’efficacia del trapianto di cornea combinato con LSCT per la GDLD, dimostrando che può prolungare l’intervallo prima della recidiva prevenendo la reinvasione delle cellule epiteliali dell’ospite 10). Successivamente, anche Movahedan et al. hanno riportato un approccio simile 11).
Per rivestire la superficie oculare con l’epitelio corneale derivato dal trapianto, si rimuove l’epitelio corneale dell’ospite e si esegue un trapianto limbare. Dopo l’intervento, si continua l’uso continuo di lenti a contatto terapeutiche per ritardare la recidiva.
Negli ultimi anni si sta valutando anche l’indicazione per la cornea artificiale (Boston tipo I Kpro). Poiché non passa attraverso l’epitelio corneale dell’ospite, teoricamente evita il riaccumulo di amiloide, ma comporta rischi di complicanze postoperatorie come infezioni e membrana retroprotesica.
QLa recidiva si verifica anche dopo il trapianto di cornea?
A
Nella GDLD, il tasso di recidiva dopo trapianto di cornea è estremamente elevato. È stato riportato che circa il 97% dei casi recidiva entro 4 anni dalla cheratoplastica perforante (PKP). La causa principale è la sostituzione dell’epitelio del trapianto da parte delle cellule epiteliali del ricevente. Come misure sviluppate in Giappone, si utilizzano la combinazione di trapianto di cellule staminali limbari corneali 10) e l’uso continuo di lenti a contatto terapeutiche 7) per ritardare la recidiva. Nella gestione a lungo termine, l’obiettivo è mantenere la funzione visiva e prolungare gli intervalli tra gli interventi, presupponendo che la recidiva si verifichi.
6. Fisiopatologia e meccanismo dettagliato della malattia
Il gene TACSTD2, responsabile della GDLD, è un gene a singolo esone situato sul cromosoma 1p32. Nel 1999, Tsujikawa e colleghi lo hanno identificato come gene causale mediante analisi di linkage in famiglie giapponesi 4). La proteina TACSTD2 svolge un ruolo indispensabile nel mantenimento della funzione di barriera dell’epitelio corneale.
Quando si verifica una mutazione con perdita di funzione nel gene TACSTD2, la normale localizzazione intracellulare delle proteine delle giunzioni strette Claudina 1 e Claudina 7 viene compromessa. Nakatsuka e colleghi hanno dimostrato, mediante analisi di cellule epiteliali corneali coltivate e di famiglie giapponesi, che la perdita di funzione di TACSTD2 porta alla perdita della localizzazione delle claudine alla giunzione apicolaterale e a una ridotta funzione di barriera epiteliale 5). Inoltre, nel 2011 hanno riportato nuove mutazioni di TACSTD2 in tre famiglie e la loro anomala localizzazione intracellulare 12).
A causa della ridotta funzione di barriera epiteliale, proteine come la lattoferrina del liquido lacrimale penetrano nella cornea. La lattoferrina penetrata forma fibrille amiloidi che si depositano sotto l’epitelio corneale. I depositi amiloidi contengono lattoferrina, ma questa malattia non è un’anomalia del gene della lattoferrina.
Istologicamente, l’opacità lattescente subepiteliale si colora in arancione-rosso con la colorazione rosso Congo e mostra birifrangenza verde mela al microscopio a luce polarizzata. Al microscopio elettronico, si osserva che le giunzioni strette dell’epitelio sono sostituite da spazi elettron-trasparenti. I depositi invadono anche le lamelle corneali, causando degenerazione delle fibre di collagene e dei proteoglicani.
Sono state riportate oltre 20 mutazioni del gene TACSTD2 12). In Giappone, la mutazione Q118X (mutazione nonsenso, nullo funzionale) è una mutazione fondatrice che rappresenta oltre l’80% dei cromosomi patogeni 2). La malattia si manifesta tipicamente negli omozigoti, ma può verificarsi anche in eterozigoti composti da matrimoni tra famiglie diverse. È interessante notare che, anche nello stesso omozigote Q118X, si osservano miste le quattro forme cliniche (a gelso, a banda, a kumquat e opacità stromale) 3).
L’amiloidosi corneale è classificata in primaria o secondaria, sistemica o locale. La GDLD, insieme alla distrofia corneale reticolare, è classificata come amiloidosi primaria locale. La degenerazione amiloide secondaria locale si verifica in associazione a trichiasi, cheratocono, traumi, uso prolungato di lenti a contatto, ecc., ed è oggetto di diagnosi differenziale.
La GDLD è una malattia rara e c’era il problema della scarsità di medici con esperienza clinica nei singoli istituti, nonché della mancanza di metodi standardizzati di diagnosi e trattamento. I criteri diagnostici e la classificazione della gravità sono stati elaborati dal gruppo di ricerca sull’epidemiologia delle malattie corneali rare e refrattarie del Ministero della Salute, del Lavoro e del Welfare e dal gruppo di ricerca per la creazione e la diffusione di linee guida cliniche per le malattie rare del segmento anteriore 2). Nel 2019, la GDLD è stata riconosciuta come malattia rara designata con il nome di «distrofia corneale a gocce gelatinose», e attualmente sono in fase di elaborazione linee guida cliniche conformi a Minds (Medical Information Network Distribution Service) 2).
A lungo termine, le recidive e i trattamenti multipli rappresentano un problema. La combinazione di lenti a contatto terapeutiche e trapianto limbare può ritardare la recidiva e prolungare l’intervallo tra gli interventi chirurgici, migliorando così la prognosi a vita 6, 7, 10).
Segnalazioni di casi atipici e recidiva unilaterale
Maeno et al. hanno riportato un caso clinicamente atipico che mostrava deposito amiloide ricorrente in un solo occhio, dimostrando la diversità fenotipica della GDLD13). Il test genetico del gene TACSTD2 gioca un ruolo determinante nella diagnosi di tali casi atipici 2).
Nella ricerca di base, ci si aspetta di chiarire in dettaglio la dinamica delle molecole delle giunzioni strette a valle di TACSTD2 e di sviluppare terapie mirate alla stabilizzazione della claudina. Dal punto di vista clinico, si sta valutando l’estensione delle indicazioni degli approcci di medicina rigenerativa, come le cornee artificiali, il trapianto di foglietti epiteliali corneali e l’epitelio corneale derivato da cellule iPS.
Ide T, Nishida K, Maeda N, Tsujikawa M, Yamamoto S, Watanabe H, et al. A spectrum of clinical manifestations of gelatinous drop-like corneal dystrophy in japan. American journal of ophthalmology. 2004;137(6):1081-4. doi:10.1016/j.ajo.2004.01.048. PMID:15183793.
Tsujikawa M, Kurahashi H, Tanaka T, Nishida K, Shimomura Y, Tano Y, et al. Identification of the gene responsible for gelatinous drop-like corneal dystrophy. Nature genetics. 1999;21(4):420-3. doi:10.1038/7759. PMID:10192395.
Nakatsukasa M, Kawasaki S, Yamasaki K, Fukuoka H, Matsuda A, Tsujikawa M, et al. Tumor-associated calcium signal transducer 2 is required for the proper subcellular localization of claudin 1 and 7: implications in the pathogenesis of gelatinous drop-like corneal dystrophy. Am J Pathol. 2010;177(3):1344-1355. PMID: 20651236. PMCID: PMC2928967. doi:10.2353/ajpath.2010.100149.
Kawasaki S, Kinoshita S. Clinical and basic aspects of gelatinous drop-like corneal dystrophy. Dev Ophthalmol. 2011;48:97-115. PMID: 21540633. doi:10.1159/000324079.
Maeno S, Soma T, Tsujikawa M, Shigeta R, Kawasaki R, Oie Y, et al. Efficacy of therapeutic soft contact lens in the management of gelatinous drop-like corneal dystrophy. The British journal of ophthalmology. 2020;104(2):241-246. doi:10.1136/bjophthalmol-2018-313809. PMID:31023713.
Hieda O, Kawasaki S, Yamamura K, Nakatsukasa M, Kinoshita S, Sotozono C. Clinical outcomes and time to recurrence of phototherapeutic keratectomy in Japan. Medicine. 2019;98(27):e16216. doi:10.1097/MD.0000000000016216. PMID:31277131; PMCID:PMC6635226.
Shimazaki J, Shimmura S, Tsubota K. Limbal stem cell transplantation for the treatment of subepithelial amyloidosis of the cornea (gelatinous drop-like dystrophy). Cornea. 2002;21(2):177-80. doi:10.1097/00003226-200203000-00010. PMID:11862090.
Nakatsukasa M, Kawasaki S, Yamasaki K, Fukuoka H, Matsuda A, Nishida K, et al. Two novel mutations of TACSTD2 found in three Japanese gelatinous drop-like corneal dystrophy families with their aberrant subcellular localization. Molecular vision. 2011;17:965-70. PMID:21541270; PMCID:PMC3084224.
Maeno S, Soma T, Nishida K. A Case of Clinically Atypical Gelatinous Drop-like Corneal Dystrophy With Unilateral Recurrent Amyloid Depositions. Cornea. 2022;41(11):1447-1450. doi:10.1097/ICO.0000000000003070. PMID:36219213.
Copia il testo dell'articolo e incollalo nell'assistente IA che preferisci.
Articolo copiato negli appunti
Apri un assistente IA qui sotto e incolla il testo copiato nella chat.