La distrofia corneale di Schnyder (Schnyder corneal dystrophy; SCD) è una distrofia corneale ereditaria causata da una mutazione del gene UBIAD1 (UbiA prenyltransferase domain containing 1), che porta a un’anomalia del metabolismo lipidico nella cornea e a un deposito anomalo di colesterolo e fosfolipidi nello stroma corneale. La trasmissione è autosomica dominante e il locus genico è situato su 1p36.
Fu riportata per la prima volta nel 1924 da Van Went e Winbaut. Dopo che l’oftalmologo svizzero Schnyder descrisse casi familiari su tre generazioni nel 1929, il nome della malattia si affermò. Nella classificazione IC3D è classificata tra le distrofie corneali stromali.
La malattia esordisce bilateralmente, con opacità corneale che inizia nell’infanzia, ma la progressione è lenta. Una caratteristica clinica è che la riduzione dell’acuità visiva è lieve rispetto al grado di opacità corneale.
QQuanto influisce sulla vista?
A
Spesso non si osserva una significativa riduzione dell’acuità visiva rispetto ai reperti alla lampada a fessura. Tuttavia, con l’età l’opacità progredisce e possono verificarsi disturbi visivi dovuti ad abbagliamento (glare) e diffusione della luce. Secondo alcuni studi, circa il 54% dei pazienti sopra i 50 anni e circa il 77% sopra i 70 anni necessitano infine di un trapianto di cornea.
Gimenez JB, Izdebska J, Szaflik JP. Schnyder Corneal Dystrophy in an Adolescent: A Case Report With Multimodal Imaging. Cureus. 2025 Aug 11; 17(8):e89786. Figure 3. PMCID: PMC12421702. License: CC BY.
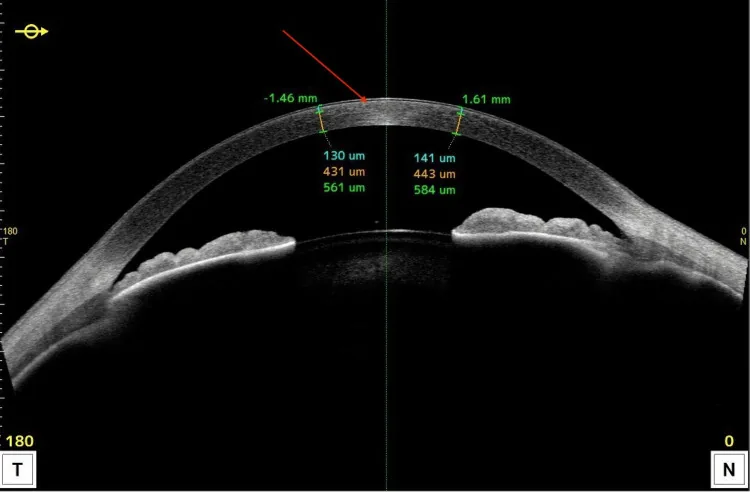
L’OCT del segmento anteriore mostra lesioni iperriflettenti localizzate nella parte anteriore della cornea e variazioni di spessore. Permette di spiegare facilmente in sezione in quale strato si distribuiscono principalmente i depositi lipidici.
Abbagliamento (glare) : causato dalla diffusione della luce da parte di cristalli o opacità nella cornea. Tende a peggiorare con l’età.
Riduzione dell’acuità visiva in condizioni di luce (visione fotopica) : a causa della dispersione della luce, la vista è particolarmente ridotta in ambienti luminosi.
Riduzione dell’acuità visiva : progredisce lentamente con l’aumentare dell’opacità, ma spesso è lieve rispetto ai segni osservati.
Segni clinici (segni rilevati dal medico durante l’esame)
I segni corneali progrediscono con l’età secondo uno schema caratteristico.
Fascia d’età
Principali segni corneali
Infanzia – adolescenza
Opacità cristallina centrale della cornea
20–30 anni
Comparsa di opacità limbare simile all’arco senile
40 anni e oltre
Opacità centrale e limbare che progredisce a tutto spessore
Cristalli corneali : presenti in circa il 50% dei casi. Si osservano come accumuli di fini cristalli aghiformi nello stroma anteriore (vicino alla membrana di Bowman).
Opacità corneale centrale : inizialmente si presenta come un’opacità grigia rotonda o ovale nello stroma superficiale. Progredendo, si estende agli strati medio e profondo.
Opacità limbare simile all’arco senile (arcus lipoides) : compare a partire dai 20 anni nella regione periferica della cornea. È caratteristica la sua comparsa in giovane età, a differenza del normale arco senile.
Zona chiara intermedia: dopo i 40 anni, l’area periferica intermedia tra l’opacità centrale e l’opacità limbale rimane relativamente trasparente.
Ipoestesia corneale: si osserva una scomparsa dei nervi corneali, ma non sono riportati casi di cheratopatia neurotrofica evidente.
In anatomia patologica, la colorazione Oil Red O colora le goccioline lipidiche in rosso. Al microscopio elettronico si osservano vacuoli nello stroma corneale.
La causa della SCD è una mutazione nel gene UBIAD1 (1p36). UBIAD1 codifica una preniltransferasi che sintetizza il menachinone-4 (MK-4, vitamina K2). Questa mutazione genetica causa un’anomalia del metabolismo lipidico corneale, con deposito di colesterolo nello stroma corneale. Il test genetico è utile per la diagnosi definitiva.
Iperlipidemia (ipercolesterolemia): può essere associata a un’alterazione sistemica del metabolismo lipidico.
Genu valgum: è nota un’associazione con anomalie scheletriche.
Malformazioni delle dita: raramente associate a malformazioni vertebrali o digitali.
QCi sono complicanze sistemiche?
A
È nota l’associazione con ipercolesterolemia e si raccomanda la valutazione del profilo lipidico. Inoltre, possono essere presenti genu valgum e, raramente, malformazioni delle dita o della colonna vertebrale.
Nonostante i reperti alla lampada a fessura, la riduzione dell’acuità visiva è spesso lieve e molti casi non richiedono un trattamento attivo. Il follow-up regolare è la base.
PTK (cheratectomia fototerapeutica con laser ad eccimeri) : eseguita per rimuovere i cristalli subepiteliali quando influenzano la vista.
Cheratoplastica lamellare profonda (DALK) : presa in considerazione nei casi avanzati in cui l’opacità si estende agli strati profondi dello stroma.
Cheratoplastica perforante (PKP) : eseguita in caso di grave opacità a tutto spessore. Secondo alcuni rapporti, circa il 54% dei pazienti sopra i 50 anni e circa il 77% sopra i 70 anni necessita infine di cheratoplastica perforante.
Attualmente non esiste una terapia farmacologica per arrestare la progressione.
QSi verifica recidiva dopo cheratoplastica?
A
Dopo cheratoplastica perforante è possibile una recidiva di depositi di colesterolo sul trapianto. I tempi e l’entità della recidiva variano da persona a persona, ma è necessario un follow-up regolare dopo il trapianto.
La SCD è causata da una mutazione nel gene UBIAD1. UBIAD1 codifica una preniltransferasi che sintetizza il menachinone-4 (vitamina K2); la disfunzione di questo enzima porta a un’alterazione del metabolismo lipidico nella cornea.
Normalmente lo stroma corneale contiene quantità molto piccole di colesterolo e fosfolipidi. Tuttavia, la mutazione UBIAD1 altera in particolare il metabolismo del colesterolo HDL, portando a un accumulo eccessivo di colesterolo e fosfolipidi nello stroma corneale. I lipidi accumulati precipitano come cristalli, compromettendo la trasparenza della cornea.
La microscopia confocale ha confermato la scomparsa dei nervi corneali. Tuttavia, nonostante la perdita nervosa, non è stata riportata una cheratopatia neurotrofica evidente. Il meccanismo con cui i depositi lipidici influenzano i nervi corneali non è completamente chiarito.