A distrofia macular da córnea (MCD) é uma distrofia corneana hereditária na qual glicosaminoglicanos (principalmente sulfato de queratana) se acumulam no estroma da córnea. Segue um padrão de herança autossômica recessiva e é causada por mutação no gene CHST6 localizado no braço longo do cromossomo 16 (16q22)1,3). Antigamente era chamada de distrofia corneana de Groenouw tipo II ou distrofia corneana de Fehr.
Ao contrário de muitas outras distrofias do estroma corneano (granular, lattice) que são autossômicas dominantes, esta doença é caracterizada por herança autossômica recessiva. No Japão, é contada como uma das quatro grandes distrofias da córnea juntamente com a distrofia granular (tipo I e II), distrofia lattice (tipo I e IIIA) e distrofia gelatinosa em gotas, que juntas representam cerca de 96% de todas as distrofias da córnea. As duas primeiras são autossômicas dominantes, enquanto as duas últimas (gelatinosa em gotas e macular) são autossômicas recessivas.
Na classificação IC3D (International Committee for Classification of Corneal Dystrophies), a MCD é posicionada como um tipo de distrofia estromal 1). Na revisão de Aggarwal et al. no Survey of Ophthalmology de 2018, esta doença é descrita como “distrofia estromal rara, mas com grande impacto na função visual”, e o sistema de diagnóstico e tratamento é resumido 4). Na 2ª edição da classificação IC3D, as distrofias corneanas são classificadas nas categorias 1 a 4 com base na força das evidências do gene causador, achados patológicos e quadro clínico 1). A MCD é classificada como categoria 1 (distrofia estabelecida em nível genético) devido à identificação da mutação no gene CHST6.
Historicamente, esta doença foi descrita pela primeira vez por Groenouw em 1890, e posteriormente surgiu a tradição de nomear a distrofia granular como “tipo I” e a distrofia macular como “tipo II”. Em 1938, Jones e Zimmerman a estabeleceram como doença independente, e em 2000, a base molecular foi elucidada por Akama et al. através da identificação do gene CHST6 3).
Globalmente, há grande variação geográfica, e em áreas de alta prevalência, observa-se agregação familiar. É uma doença relativamente rara.
Variação Geográfica da Prevalência
Estados Unidos: cerca de 0,3 por 250.000 pessoas, raro 2,3)
Islândia: cerca de 19 por 250.000 pessoas, uma das áreas de maior frequência no mundo 5,6)
Áreas de alta prevalência: Sul da Índia, Arábia Saudita, Islândia e Escandinávia 5,7)
Outras áreas: relativamente raro. Ocorre em casamentos consanguíneos ou heterozigotos compostos
Fenótipo Imune
Tipo I: negativo para sulfato de queratana na córnea e soro 2)
Tipo IA: positivo nos ceratócitos da córnea, negativo no soro 2)
Tipo II: positivo para sulfato de queratana na córnea e soro 2)
Quadro clínico: O fenótipo dos três tipos é idêntico e indistinguível à lâmpada de fenda2,8)
O fenótipo imune da MCD é classificado com base na quantidade de sulfato de queratana na córnea e soro usando anticorpos monoclonais anti-sulfato de queratana 2,3).
Fenótipo
Queratano sulfato da córnea
Queratano sulfato sérico
Tipo I
Negativo
Negativo
Tipo IA
Positivo (intracelular)
Negativo
Tipo II
Positivo
Positivo
A maioria dos pacientes é classificada como tipo I ou IA. No entanto, clinicamente, a distinção entre esses subtipos não é importante e não pode ser diferenciada pelo exame2,8).
QQual a diferença entre a distrofia macular da córnea e outras distrofias da córnea?
A
A maior diferença é que segue herança autossômica recessiva. Enquanto as distrofias corneanas granular e reticular são autossômicas dominantes, esta doença requer mutações em ambos os alelos do gene CHST6. Além disso, apresenta opacidade difusa em vidro fosco, embaçamento de toda a córnea e, ao exame com lâmpada de fenda, depósitos nas camadas superficiais no centro e nas camadas profundas na periferia.
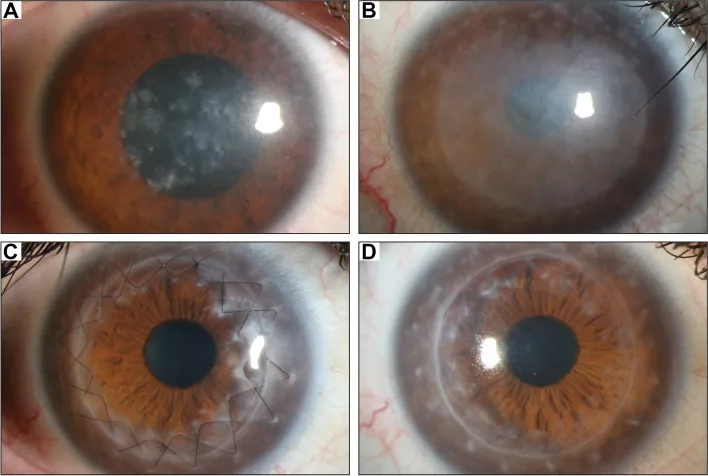
Gassel CJ, et al. Histological findings of corneal tissue after failed phototherapeutic keratectomy in macular corneal dystrophy - a case report. BMC Ophthalmol. 2022. Figure 1. PMCID: PMC9080147. License: CC BY.
Fotografia com lâmpada de fenda mostrando opacidade difusa acinzentada e depósitos maculares do centro para toda a córnea. Mostra os achados clínicos típicos da distrofia macular da córnea, facilitando a compreensão da opacidade corneana que causa diminuição da visão.
O padrão típico de progressão dos achados clínicos é o seguinte.
Clinicamente, depósitos finos difusos são vistos no estroma corneano, causando opacidade em vidro fosco. Com a progressão, a opacidade se estende por toda a espessura do estroma e se espalha do centro para a periferia. Posteriormente, além da opacidade leve, muitas opacidades pequenas e irregulares de cor branco-acinzentada são vistas no estroma superficial a profundo.9)
Achados iniciais
Opacidade puntiforme: Pequenas opacidades puntiformes branco-acinzentadas aparecem no estroma superficial da córnea central
Opacidade em vidro fosco: Opacidade difusa leve é observada no estroma corneano
Bordas mal definidas: As margens da opacidade são indistintas, e o limite com o estroma normal é pouco nítido
Sinais de Estágio Avançado
Extensão para todas as camadas: A opacidade se estende por toda a espessura do estroma
Extensão para a periferia: A opacidade se espalha do centro para a periferia
Afinaçãocorneana: A espessura da córnea central diminui
Depósitos no endotélio e membrana de Descemet: Substâncias anormais também se acumulam nas estruturas profundas
Na lâmpada de fenda, toda a córnea apresenta opacidade difusa com depósitos irregulares acinzentados. Ao fazer um corte óptico com a fenda de luz, observa-se uma distribuição característica: os depósitos localizam-se nas camadas superficiais na região central e nas camadas profundas na periferia. As lesões em placas frequentemente aparecem em padrão circular concêntrico8).
A opacidade pode se estender ao limbo, o que é um importante ponto de diferenciação de outras distrofias corneanas. Na distrofia corneana granular e na distrofia corneana lattice, o limbo frequentemente permanece transparente, enquanto na MCD, toda a córnea frequentemente se torna opaca até o limbo2,8). Além disso, o aparecimento de astigmatismo irregular está relacionado aos depósitos no estroma anterior, e pode ocorrer diminuição da sensibilidade corneana. Devido aos depósitos de substâncias anormais também no endotélio, em casos avançados pode ocorrer edema estromal por disfunção endotelial8).
A história natural varia entre indivíduos, mas frequentemente segue os seguintes estágios.
Infância (fase assintomática): A mutação genética está presente desde o nascimento, mas os achados à lâmpada de fenda são escassos e assintomáticos
Idade escolar à adolescência (fase de opacidade inicial): Opacidade difusa leve aparece nas camadas superficiais do estroma corneano, e posteriormente depósitos em placas são observados
10-30 anos (fase de declínio visual): A opacidade progride e o paciente começa a perceber a diminuição da visão
30-40 anos (fase avançada): A opacidade se espalha por toda a espessura do estroma e limbo, e a diminuição da sensibilidade corneana, afinamento corneano e astigmatismo irregular tornam-se evidentes
Meia-idade em diante (fase de indicação de transplante de córnea): A diminuição da função visual atinge um nível que interfere nas atividades diárias, e o transplante é considerado
A MCD é uma doença progressiva na qual o declínio da função visual continua ao longo da vida, diferindo da distrofia corneana granular tipo I e de alguns subtipos de distrofia corneana lattice, onde o comprometimento visual é leve4,8).
O gene causador é CHST6 (carbohydrate sulfotransferase 6)3). Localizado em 16q22, codifica uma enzima que transfere grupos sulfato para N-acetilglicosamina em proteoglicanos da córnea. Os tipos de mutações neste gene são muito variados, incluindo mutações de sentido trocado, sem sentido, de deslocamento de quadro e deleções na região 5’ a montante, relatadas em diferentes grupos étnicos 3,7).
Como a herança é autossômica recessiva, geralmente ambos os pais do probando são portadores. A incidência tende a ser maior em regiões ou grupos com alta frequência de casamentos consanguíneos. A doença também pode ocorrer em heterozigotos compostos resultantes de casamentos entre famílias diferentes 7).
Akama et al. em 2000 identificaram o CHST6 como o gene causador desta doença e mostraram que tanto o fenótipo imune tipo I quanto o tipo II são causados por mutações no mesmo locus gênico 3). Esta descoberta foi importante, sugerindo que a diferença no fenótipo imune é determinada por diferentes padrões de mutação em um único gene, e formou a base para o sistema de diagnóstico genético subsequente.
Mais de 200 tipos de mutações foram relatados, sendo as mutações de sentido trocado as mais comuns. Sultana et al. identificaram várias mutações novas em um grupo de pacientes no sul da Índia e mostraram que a alta frequência naquela região se deve ao acúmulo de mutações específicas da região 7). Acúmulos regionais semelhantes foram relatados na Arábia Saudita e Islândia, e acredita-se que o histórico populacional (efeito fundador e costumes de casamentos consanguíneos) contribua para a frequência de ocorrência 5,6,7).
A frequência de ocorrência varia conforme a região, sendo uma doença relativamente rara 9). Comparada a outras distrofias corneanas, como a distrofia corneana granular, o número de casos é menor, e tende a ser relatada em famílias com histórico de consanguinidade ou heterozigotos compostos.
Histórico familiar: Como a herança é autossômica recessiva, ambos os pais precisam ser portadores
Casamento consanguíneo: Aumenta a taxa de incidência
Fator geográfico: Alta prevalência no sul da Índia, Arábia Saudita, Islândia e Escandinávia 5,7)
QO teste genético é necessário?
A
O teste do gene CHST6 é útil para o diagnóstico definitivo. O teste genético pode ser realizado em instituições médicas credenciadas. No entanto, em muitos serviços, o diagnóstico clínico é baseado principalmente no exame com lâmpada de fenda. O teste genético é útil para avaliar o risco em futuros filhos e para confirmar o diagnóstico em casos com achados clínicos atípicos.
A suspeita de MCD surge em adolescentes a adultos jovens com opacidade difusa de toda a córnea, bilateral e progressiva. Primeiro, obtém-se história detalhada dos sintomas subjetivos (baixa visual, fotofobia, irritação), história familiar e consanguinidade. Em seguida, realiza-se avaliação da córnea com lâmpada de fenda, avaliação da função endotelial e, se necessário, teste genético.
Este é o exame básico para o diagnóstico. Se houver opacidade corneana bilateral sem hiperemia ou edema, suspeite de distrofia corneana. Na MCD, os seguintes achados são característicos:
Opacidade difusa em vidro fosco: Toda a córnea torna-se opaca difusamente
Distribuição concêntrica: Ao corte com feixe de luz, camadas superficiais no centro e profundas na periferia
Infiltração límbica: A opacidade pode se estender até o limbo
Anormalidade da superfície endotelial: Em casos avançados, podem ser observados depósitos gutatos
Enquanto a maioria das distrofias corneanas é observada ao nível da fenda como lesões descontínuas (com áreas transparentes entre os depósitos), a MCD excepcionalmente apresenta padrão de opacidade difusa. Juntamente com a distrofia corneana reticular tipo I e a distrofia corneana gelatinosa em gotas, é um exemplo típico de “depósitos corneanos observados difusamente”.
Microscopia Confocal (in vivo confocal microscopy): Mostra material altamente refletivo com bordas mal definidas e perda de imagens de ceratócitos normais8)
Topografia Corneana: Mostra aumento da densidade no ápice corneano e afinamento corneano central
Microscopia Ultrassônica Biomicroscópica (UBM): Útil para avaliar opacidades profundas e estruturas posteriores da córnea
Microscopia Especular: Avaliação da densidade e morfologia das células endoteliais. O grau de lesão endotelial está diretamente relacionado à escolha da técnica cirúrgica
Pentacam Corneano (Câmera Scheimpflug): Fornece um mapa de densidade de todas as camadas da córnea, útil para avaliação tridimensional de áreas de opacidade
Histologicamente, a coloração com azul alciano e ferro coloidal mostra positividade, e observa-se acúmulo difuso de glicosaminoglicanos hipossulfatados intra e extracelularmente nos ceratócitos do estroma corneano2,8). Podem ser observadas rupturas na membrana de Bowman e, em casos avançados, material anormal também é encontrado dentro das células endoteliais. Podem ocorrer achados semelhantes a guttae na membrana de Descemet.
Presente desde o nascimento, espessamento estromal
Mucopolissacaridose sistêmica (tipo corneano)
Recessiva/ligada ao X
Acompanhada de sintomas sistêmicos
Além disso, a distrofia corneana posterior amorfa (PACD) e a distrofia corneana pré-Descemet (PDCD) também entram no diagnóstico diferencial. A mucopolissacaridose sistêmica (como síndromes de Hurler, Scheie, Morquio) também pode causar opacidade corneana, portanto, deve-se avaliar incluindo achados sistêmicos8).
QComo é diagnosticada a distrofia corneana macular?
A
O diagnóstico clínico é feito com lâmpada de fenda mostrando opacidade corneana difusa em vidro fosco e depósitos maculares, além de bilateralidade, progressão, história familiar e idade de início (10-30 anos). O teste genético CHST6 é útil para confirmação.
Os objetivos do tratamento para MCD são: (1) manter ou restaurar a função visual, (2) aliviar a dor e os sintomas de irritação estabilizando a superfície ocular, (3) prevenir complicações (erosão epitelial e infecção). Como não existe terapia causal fundamental, a intervenção é realizada gradualmente de acordo com o estágio e os sintomas.
Em casos com declínio visual progressivo, o transplante de córnea é o único tratamento curativo. A técnica é escolhida com base na presença ou ausência de doença endotelial.
Ceratoplastia lamelar anterior profunda (DALK): É a primeira escolha em casos sem envolvimento endotelial 8,9). Por preservar o endotélio da própria córnea, o risco de rejeição do enxerto é menor do que na ceratoplastia penetrante. No relatório de avaliação da AAO (Academia Americana de Oftalmologia), a DALK para distrofia estromal foi avaliada como proporcionando recuperação da função visual equivalente à PKP com menor perda endotelial 8). A taxa de recorrência após DALK é baixa, e a recorrência é rara porque o estroma do receptor é substituído.
Ceratoplastia penetrante (PKP): Indicada em casos avançados com envolvimento endotelial ou da membrana de Descemet com depósitos de substâncias anormais, e em casos com afinamento central grave da córnea7). A idade média para a primeira PKP por MCD é de 30 a 40 anos 7), e a taxa de sobrevivência do enxerto é boa.
Ceratectomia fototerapêutica (PTK): Realizada sintomaticamente para erosão epitelial recorrente ou opacidade cicatricial superficial. No entanto, deve-se atentar para a hipermetropização e indução de opacidade estromal.
Na MCD, substâncias anormais também podem se depositar nas células endoteliais, portanto, em casos com doença que se estende profundamente, a PKP tende a ser escolhida em vez da DALK7). Na presença de anormalidade endotelial, a ceratoplastia penetrante é indicada. A avaliação endotelial pré-operatória detalhada (microscopia especular ou confocal) é fundamental para determinar a técnica cirúrgica.
A taxa de recorrência após DALK é relatada como baixa, em torno de 5%. Enquanto a taxa de sobrevivência do enxerto após PKP é boa, com muitos relatos de sobrevivência a longo prazo, alguns casos relatam redeposição de material anormal no enxerto após vários anos a décadas de cirurgia8). Na série de Al-Swailem et al. na Arábia Saudita, a taxa de sobrevivência do enxerto após PKP para MCD foi boa, mas a recorrência foi observada em alguns casos durante o acompanhamento de longo prazo8). Além disso, na revisão da AAO por Reinhart et al., foi demonstrado que o DALK alcança recuperação visual igual ou superior ao PKP com baixa taxa de perda de células endoteliais9). O estudo multicêntrico de Unal et al. também relatou a eficácia do DALK em distrofias estromais incluindo MCD.
Geralmente, a recorrência pós-transplante de córnea ocorre porque a patologia molecular da doença original permanece no hospedeiro. No DALK, o endotélio do hospedeiro e a camada anterior da membrana de Descemet são preservados, portanto, deve-se notar que a doença pode progredir em casos com lesões endoteliais. Após a cirurgia, é necessário acompanhamento periódico de longo prazo (exame de acuidade visual, lâmpada de fenda, medição da densidade de células endoteliais).
QQual deve ser escolhido, DALK ou PKP?
A
Se o endotélio ou a membrana de Descemet não estiverem envolvidos, o transplante de camada anterior profunda (DALK) é a primeira escolha. O DALK preserva o endotélio da própria córnea, reduzindo o risco de rejeição, e a taxa de recorrência é relatada em cerca de 5% após a cirurgia. Por outro lado, em casos onde há depósitos de material anormal também no endotélio, o transplante de córnea de espessura total (PKP) é indicado. A técnica cirúrgica é determinada com base na avaliação endotelial pré-operatória.
6. Fisiopatologia e Mecanismo Detalhado de Ocorrência
O gene CHST6 codifica a carboidrato sulfotransferase 6 (carbohydrate sulfotransferase 6)3). Esta enzima é responsável pela transferência do grupo sulfato para a N-acetilglicosamina na molécula de queratana, e é essencial para a síntese normal do sulfato de queratana (KS) contido nos proteoglicanos da córnea.
Quando a atividade enzimática é perdida devido a uma mutação genética, é sintetizado sulfato de queratana hipossulfatado e incompletamente sulfatado. Este sulfato de queratana anormal tem baixa solubilidade e se deposita anormalmente dentro e fora dos ceratócitos no estroma da córnea2,3).
As anormalidades quantitativas e qualitativas do sulfato de queratana levam à seguinte cascata patológica.
Produção anormal de pequenos proteoglicanos ricos em leucina (SLRP): SLRPs específicos da córnea, como lumican, keratocan e mimecan, não são sintetizados normalmente
Anormalidade no arranjo das fibras de colágeno: Esses SLRPs controlam rigorosamente o diâmetro das fibras de colágeno da córnea e o espaçamento entre elas, garantindo a transparência. A diminuição da função dos SLRPs resulta em diâmetro não uniforme das fibras de colágeno e alteração no espaçamento entre elas 2)
Acúmulo anormal na matriz extracelular: O próprio queratano não sulfatado se deposita na matriz extracelular
Aumento da dispersão da luz e perda de transparência: As alterações combinadas acima aumentam a dispersão da luz visível, causando opacidade difusa em toda a córnea
O acúmulo de glicosaminoglicanos é observado dentro e fora dos ceratócitos do estroma e, com a progressão da doença, se espalha para a membrana de Bowman, membrana de Descemet e células endoteliais. No tipo I, a diminuição da atividade enzimática também foi confirmada na cartilagem auricular, sugerindo a possibilidade de ser parte de um distúrbio metabólico sistêmico do sulfato de queratano 2). No entanto, os sintomas sistêmicos raramente se manifestam clinicamente, sendo tratado como uma doença localizada com sintomas principais na córnea.
Entre os proteoglicanos da córnea, o lumican tem o papel de controlar o diâmetro das fibras de colágeno para cerca de 25 nm, enquanto o keratocan e o mimecan mantêm o espaçamento uniforme entre as fibras. Quando as cadeias laterais sulfatadas desses SLRPs são curtas e incompletas, as fibras de colágeno apresentam variação na espessura e o espaçamento entre elas se torna irregular. Como resultado, a dispersão da luz dentro do estroma da córnea aumenta, sendo observada clinicamente como uma opacidade semelhante a vidro fosco.
A teoria reticular de Maurer é classicamente conhecida para a manutenção da transparência da córnea através do “cancelamento da interferência da luz pelo arranjo reticular ordenado das fibras de colágeno”, mas na MCD, essa estrutura reticular é rompida devido à anormalidade dos SLRPs, resultando em perda de transparência 2,4).
Em pesquisas básicas recentes, foi relatado que a disfunção da autofagia (autofagia) causada pela mutação CHST6 pode induzir piroptose (morte celular inflamatória) dos ceratócitos, contribuindo para a progressão da doença 8). Anormalidades semelhantes de autofagia também foram relatadas em outras distrofias corneanas (como o tipo granular II), chamando a atenção como um mecanismo patológico comum das distrofias corneanas em geral.
A terapia genética direcionada foi proposta como uma estratégia de tratamento permanente 8). Pesquisas básicas sobre edição genética usando CRISPR/Cas9 estão em andamento na distrofia epitelial da córnea de Meesmann, e pode se tornar uma opção de tratamento futura para a MCD. No entanto, ainda existem muitos desafios para a aplicação clínica, como edição não intencional de alelos normais (efeitos fora do alvo), desenvolvimento de métodos eficientes de entrega de genes para células do estroma corneano e verificação de segurança a longo prazo.
Uma abordagem para remover enzimaticamente o sulfato de queratana hipossulfatado acumulado na córnea está sendo investigada em nível básico. Isso aplica o conceito de terapia de reposição enzimática usada na mucopolissacaridose sistêmica localmente na córnea, mas não há relatos de aplicação clínica até o momento.
A ceratoplastia endotelial automatizada com remoção da membrana de Descemet (DSAEK) e a ceratoplastia endotelial da membrana de Descemet (DMEK) são técnicas desenvolvidas principalmente para distrofia endotelial de Fuchs e ceratopatia bolhosa, mas sua aplicação em casos de MCD com envolvimento endotelial precoce é um tópico de pesquisa futura. Atualmente, como a doença afeta tanto o estroma quanto o endotélio, a ceratoplastia penetrante (PKP) é a opção realista. No futuro, o conceito de “ceratoplastia sequencial” combinando ceratoplastia lamelar anterior profunda (DALK) para substituir o estroma e DMEK para substituir o endotélio em estágios foi proposto, mas todos ainda estão em fase de pesquisa.
O nível sérico de sulfato de queratana é relatado como mais baixo que o normal em pacientes com MCD do fenótipo imunológico tipo II, e sua utilidade como marcador metabólico sistêmico está sendo discutida 2). No futuro, espera-se que seja usado para triagem por exames de sangue e diagnóstico precoce de portadores de mutações em famílias com CHST6. Além disso, a análise automatizada de imagens de lâmpada de fenda usando inteligência artificial também pode contribuir para a detecção precoce de doenças raras como esta.
Como a doença é autossômica recessiva, aproximadamente 25% dos irmãos de um paciente podem ser afetados. Usando o teste do gene CHST6, é possível realizar triagem familiar e aconselhamento genético sobre casamento e gravidez. O encaminhamento precoce a um especialista é recomendado em famílias com histórico de casamento consanguíneo ou casos de opacidade corneana semelhante na família.
Akama TO, Nishida K, Nakayama J, et al. Macular corneal dystrophy type I and type II are caused by distinct mutations in a new sulphotransferase gene. Nature Genetics. 2000;26(2):237-241. doi:10.1038/79987.
Aggarwal S, Peck T, Golen J, Karcioglu ZA. Macular corneal dystrophy: A review. Survey of Ophthalmology. 2018;63(5):609-617. doi:10.1016/j.survophthal.2018.03.004.
Jonasson F, Oshima E, Thonar EJ, et al. Macular corneal dystrophy in Iceland. A clinical, genealogic, and immunohistochemical study of 28 patients. Ophthalmology. 1996;103(7):1111-1117. doi:10.1016/s0161-6420(96)30559-9.
Jonasson F, Johannsson JH, Garner A, Rice NS. Macular corneal dystrophy in Iceland. Eye (London, England). 1989;3 ( Pt 4):446-54. doi:10.1038/eye.1989.66. PMID:2606219.
Sultana A, Sridhar MS, Jagannathan A, et al. Novel mutations of the carbohydrate sulfotransferase-6 (CHST6) gene causing macular corneal dystrophy in India. Mol Vis. 2003;9:730-734. PMID:14735064.
Al-Swailem SA, Al-Rajhi AA, Wagoner MD. Penetrating keratoplasty for macular corneal dystrophy. Ophthalmology. 2005;112(2):220-224. doi:10.1016/j.ophtha.2004.08.017.
Reinhart WJ, Musch DC, Jacobs DS, Lee WB, Kaufman SC, Shtein RM. Deep anterior lamellar keratoplasty as an alternative to penetrating keratoplasty a report by the american academy of ophthalmology. Ophthalmology. 2011;118(1):209-218. doi:10.1016/j.ophtha.2010.11.002. PMID:21199711.
Copie o texto do artigo e cole no assistente de IA de sua preferência.
Artigo copiado para a área de transferência
Abra um assistente de IA abaixo e cole o texto copiado na conversa.