Maküler kornea distrofisi (macular corneal dystrophy: MCD), kornea stromasında glikozaminoglikanların (başlıca keratan sülfat) biriktiği kalıtsal bir kornea distrofisidir. Otozomal resesif kalıtım gösterir ve nedeni kromozom 16’nın uzun kolunda (16q22) yer alan CHST6 genindeki mutasyondur1,3). Eskiden Groenouw kornea distrofisi tip II veya Fehr kornea distrofisi olarak da adlandırılırdı.
Diğer birçok kornea stroma distrofisi (granüler ve latis) otozomal dominant kalıtım gösterirken, bu hastalığın otozomal resesif kalıtım göstermesi karakteristik bir özelliktir. Japonya’da granüler kornea distrofisi (tip I ve II), latis kornea distrofisi (tip I ve IIIA) ve jelatinöz damla benzeri kornea distrofisi ile birlikte dört büyük kornea distrofisi arasında sayılır ve bunlar tüm kornea distrofilerinin yaklaşık %96’sını oluşturur. Bunlardan ilk ikisi (granüler ve latis) otozomal dominant, son ikisi (jelatinöz damla benzeri ve maküler) ise otozomal resesiftir.
IC3D (Uluslararası Kornea Distrofileri Sınıflandırma Komitesi) sınıflandırmasına göre MCD, stromal distrofilerin bir türü olarak kabul edilir 1). Aggarwal ve arkadaşlarının Survey of Ophthalmology 2018’deki derlemesinde bu hastalık, “nadir ancak görsel işlev üzerinde büyük etkisi olan bir stromal distrofi” olarak detaylandırılmış ve tanı-tedavi sistemi özetlenmiştir 4). IC3D sınıflandırmasının ikinci baskısında kornea distrofileri kategori 1-4’e ayrılır ve neden olan gen, patolojik bulgular ve klinik görünümün kanıt gücüne göre sınıflandırılır 1). MCD, CHST6 gen mutasyonunun tanımlanmasıyla kategori 1 (gen düzeyinde kanıtlanmış distrofi) olarak sınıflandırılmıştır.
Hastalığın tarihsel arka planına bakıldığında, ilk kez 1890’da Groenouw tarafından tanımlanmış ve daha sonra granüler distrofi “tip I”, maküler distrofi ise “tip II” olarak adlandırılmıştır. 1938’de Jones ve Zimmerman tarafından bağımsız bir hastalık olarak kabul edilmiş ve 2000’de Akama ve arkadaşlarının CHST6 genini tanımlamasıyla moleküler temel aydınlatılmıştır 3).
Dünya genelinde bölgesel farklılıklar büyüktür ve prevalansın yüksek olduğu bölgelerde aile içi birikim görülür. Nispeten nadir bir hastalıktır.
Prevalansta Bölgesel Farklılıklar
ABD: 250.000 kişide yaklaşık 0,3 kişi, nadirdir 2,3)
İzlanda: 250.000 kişide yaklaşık 19 kişi, dünyada en sık görülen bölgelerden biri 5,6)
Yüksek prevalanslı bölgeler: Güney Hindistan, Suudi Arabistan, İzlanda ve İskandinav ülkeleri 5,7)
Diğer bölgeler: Nispeten nadir. Akraba evlilikleri ve bileşik heterozigotlukta ortaya çıkar
İmmün Fenotip
Tip I: Kornea ve serumda keratan sülfat negatif 2)
Tip IA: Kornea keratositlerinde pozitif, serumda negatif 2)
Tip II: Kornea ve serumda keratan sülfat pozitif 2)
Klinik görünüm: Her üç tip de aynı fenotipe sahiptir ve yarık lamba ile ayırt edilemez 2,8)
MCD’nin immün fenotipi, anti-keratan sülfat monoklonal antikoru kullanılarak kornea ve serumdaki keratan sülfat miktarına göre sınıflandırılır 2,3).
Fenotip
Kornea keratan sülfat
Serum keratan sülfat
Tip I
Negatif
Negatif
Tip IA
Pozitif (hücre içi)
Negatif
Tip II
Pozitif
Pozitif
Çoğu hasta Tip I veya Tip IA olarak sınıflandırılır. Ancak klinik olarak bu alt tiplerin ayrımı önemli değildir ve muayene bulgularıyla ayırt edilemezler2,8).
QMaküler kornea distrofisi diğer kornea distrofilerinden nasıl farklıdır?
A
En büyük fark, otozomal resesif kalıtım göstermesidir. Granüler ve latis kornea distrofileri otozomal dominant kalıtılırken, bu hastalık CHST6 geninin her iki allelinde mutasyon gerektirir. Ayrıca yaygın buzlu cam benzeri bulanıklık gösterir ve tüm kornea beyazlaşır; yarık lambada merkezde yüzeysel, çevrede derin tabakalarda birikim görülmesi de karakteristiktir.
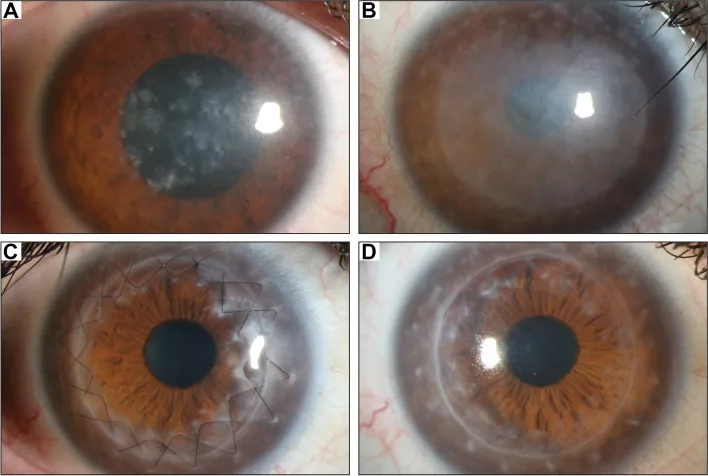
Gassel CJ, et al. Histological findings of corneal tissue after failed phototherapeutic keratectomy in macular corneal dystrophy - a case report. BMC Ophthalmol. 2022. Figure 1. PMCID: PMC9080147. License: CC BY.
Yarık lamba fotoğrafında, korneanın merkezinden tümüne kadar grimsi-beyaz yaygın bulanıklık ve lekesel birikintiler izleniyor. Maküler kornea distrofisinin tipik klinik bulgularını gösterir ve görme azalmasına neden olan kornea bulanıklığının anlaşılmasını kolaylaştırır.
Klinik bulguların tipik ilerleme paterni aşağıdaki gibidir:
Klinik olarak kornea stromasında yaygın ince birikintiler görülür ve kornea buzlu cam görünümü alır. Bulanıklık ilerledikçe stromanın tüm katmanlarına yayılır ve merkezden çevreye doğru genişler. Daha sonra hafif bulanıklığa ek olarak, stromanın yüzeyel ve derin katmanlarında çok sayıda küçük, düzensiz şekilli grimsi-beyaz bulanıklıklar ortaya çıkar.9)
Erken bulgular
Lekesel bulanıklıklar: Kornea merkezinin yüzeyel stromasında küçük grimsi-beyaz lekesel bulanıklıklar ortaya çıkar
Buzlu cam görünümünde bulanıklık: Kornea stromasında yaygın hafif bulanıklık izlenir
Sınırların belirsiz olması: Bulanıklığın kenarları nettir ve normal stroma ile sınırı belirgin değildir
İleri evre bulguları
Tüm katmanlara yayılım: Bulanıklık stromanın tüm kalınlığına ulaşır
Perifere yayılım: Bulanıklık merkezden çevreye doğru genişler
Kornea incelmesi: Santral kornea kalınlığı azalır
Endotel ve Descemet membranında birikim: Derin yapılarda da anormal maddeler birikir
Yarık lamba mikroskobunda, korneanın tamamı diffüz olarak bulanıktır ve içinde gri-beyaz düzensiz lekesel birikimler görülür. Yarık ışıkla optik kesit alındığında, merkezde birikimler yüzeyel tabakalarda, periferde ise derin tabakalarda yer alır şeklinde karakteristik bir dağılım gösterir. Lekesel lezyonlar sıklıkla eşmerkezli daireler şeklinde ortaya çıkar8).
Bulanıklık limbusa kadar uzanabilir ve bu, diğer kornea distrofilerinden önemli bir ayırt edici noktadır. Granüler kornea distrofisi ve latis kornea distrofisinde limbus genellikle saydam kalırken, MCD’de tüm kornea limbusa kadar sıklıkla bulanıktır2,8). Ayrıca, düzensiz astigmatizmanın ortaya çıkışı ön stromal birikimlerle ilişkilidir ve kornea duyusunda azalma da görülebilir. Endotelde de anormal maddeler biriktiğinden, ilerlemiş olgularda endotel fonksiyon bozukluğuna bağlı stromal ödem gelişebilir8).
Doğal seyir kişiden kişiye değişmekle birlikte, sıklıkla aşağıdaki aşamalar izlenir.
Erken çocukluk (asemptomatik dönem): Genetik mutasyon doğumdan itibaren mevcuttur, ancak yarık lamba bulguları azdır ve hasta asemptomatiktir
Okul çağı-ergenlik (erken bulanıklık dönemi): Kornea stromasının yüzeyel tabakalarında diffüz hafif bulanıklık ortaya çıkar ve zamanla lekesel birikimler görülür
10-30 yaş (görme azalması dönemi): Bulanıklık ilerler ve hasta görme azalmasını fark eder
30-40 yaş (ileri evre): Bulanıklık stromanın tüm katmanlarına ve limbusa yayılır; kornea duyusunda azalma, kornea incelmesi ve düzensiz astigmatizma belirginleşir
Orta-ileri yaş (kornea nakli endikasyonu dönemi): Görme fonksiyonundaki azalma günlük yaşamı etkileyecek düzeye ulaşır ve nakil ameliyatı düşünülür
MCD ilerleyici bir hastalıktır ve yaşam boyu görme fonksiyonunda azalma devam eder; bu yönüyle, görme bozukluğunun hafif kaldığı granüler kornea distrofisi tip I ve latis kornea distrofisinin bazı alt tiplerinden prognozu büyük ölçüde farklıdır4,8).
Sorumlu gen CHST6 (karbonhidrat sülfotransferaz 6)‘dır3). 16q22’de yer alır ve kornea proteoglikanlarındaki N-asetilglukozamine sülfat grubu aktaran bir enzimi kodlar. Bu gendeki mutasyon türleri çok fazladır ve farklı etnik gruplarda missense, nonsense, çerçeve kayması mutasyonları ve 5’ üst bölge delesyonları rapor edilmiştir3,7).
Otozomal resesif kalıtım gösterdiğinden, genellikle hastanın her iki ebeveyni de taşıyıcıdır. Akraba evliliğinin sık olduğu bölge ve toplumlarda görülme sıklığı özellikle yüksektir. Farklı aileler arasındaki evliliklerden doğan bileşik heterozigotlarda da hastalık ortaya çıkabilir7).
Akama ve ark. 2000 yılında CHST6’yı bu hastalığın sorumlu geni olarak tanımlamış ve immün fenotip tip I ve II’nin her ikisinin de aynı gen lokusundaki mutasyonlardan kaynaklandığını göstermiştir3). Bu bulgu, immün fenotip farklılıklarının tek bir gendeki farklı mutasyon paternleri tarafından belirlendiğini düşündüren önemli bir keşiftir ve sonraki genetik tanı sistemlerinin temelini oluşturmuştur.
Rapor edilen mutasyon türleri 200’ün üzerindedir ve en sık missense mutasyonları görülür. Sultana ve ark. Güney Hindistanlı hasta grubunda çok sayıda yeni mutasyon tanımlamış ve bu bölgedeki yüksek sıklığın bölgesel mutasyon birikiminden kaynaklandığını göstermiştir7). Suudi Arabistan ve İzlanda’da da benzer bölgesel birikimler rapor edilmiş olup, popülasyonların tarihsel geçmişinin (kurucu etkisi ve akraba evliliği alışkanlıkları) hastalık sıklığına katkıda bulunduğu düşünülmektedir5,6,7).
Hastalığın görülme sıklığı bölgeye göre değişir ve nispeten nadir bir hastalıktır9). Granüler kornea distrofisi gibi diğer kornea distrofilerine kıyasla daha az vaka bildirilmiştir ve genellikle bileşik heterozigot veya akraba evliliği öyküsü olan ailelerden rapor edilme eğilimindedir.
Aile öyküsü: Otozomal resesif kalıtım nedeniyle her iki ebeveynin de taşıyıcı olması gerekir
Akraba evliliği: Görülme sıklığını artırır
Bölgesellik: Güney Hindistan, Suudi Arabistan, İzlanda ve İskandinav ülkelerinde prevalans yüksektir5,7)
QGenetik test gerekli midir?
A
Kesin tanı için CHST6 gen testi faydalıdır. Genetik test, akredite sağlık kuruluşlarında yapılabilir. Ancak birçok merkezde yarık lamba mikroskobu ile klinik tanı ana yöntemdir. Gelecekteki çocukların hastalık riskini değerlendirmede ve klinik bulguları tipik olmayan vakalarda kesin tanı koymada faydalıdır.
MCD’den şüphelenmek için ipucu “çift taraflı, ilerleyici, ergenlik- genç erişkin dönemde tüm korneada yaygın bulanıklık”tır. Öncelikle semptomlar (görme azalması, fotofobi, irritasyon hissi) seyri, aile öyküsü ve akraba evliliği ayrıntılı olarak sorgulanır. Ardından yarık lamba mikroskobu ile kornea bulgularının değerlendirilmesi, endotel fonksiyon değerlendirmesi ve gerektiğinde genetik test yapılması standart yaklaşımdır.
Limbus tutulumu : Bulanıklık limbus kadar uzanabilir
Endotel yüzey anormalliği : İlerlemiş olgularda damla şeklinde birikintiler görülebilir
Birçok kornea distrofisi yarık lamba seviyesinde kesintili (birikintiler arasında saydam alanlar olan) lezyonlar olarak gözlenirken, MCD istisnai olarak yaygın bulanıklık paterni gösterir. Lattice kornea distrofisi tip I ve jelatinöz damla benzeri kornea distrofisi ile birlikte “yaygın olarak gözlenen kornea birikintileri”nin tipik örnekleri olarak tanımlanır.
Ultrasonik Biyomikroskopi (UBM): Derin opasitelerin ve kornea arka yapılarının değerlendirilmesinde faydalıdır
Speküler Mikroskop: Endotel hücre yoğunluğu ve morfolojisinin değerlendirilmesi. Endotel tutulumunun derecesi cerrahi yöntem seçimini doğrudan etkiler
Kornea Pentacam (Scheimpflug Görüntüleme Cihazı): Korneanın tüm katmanlarının yoğunluk haritasını sağlar ve opasite bölgesinin üç boyutlu değerlendirilmesinde faydalıdır
Histolojik olarak Alcian mavisi ve kolloidal demir boyaması pozitiftir ve kornea stromasındaki keratositlerin içinde ve dışında yaygın olarak düşük sülfatlı glikozaminoglikan birikimi gözlenir2,8). Bowman membranında yırtıklar görülebilir ve ilerlemiş vakalarda endotel hücrelerinde de anormal madde bulunur. Descemet membranında guttae benzeri bulgular da görülebilir.
Bunun yanı sıra, posterior polimorf kornea distrofisi (PPKD) ve pre-Descemet kornea distrofisi (PDKD) de ayırıcı tanıda yer alır. Sistemik mukopolisakkaridoz (Hurler, Scheie, Morquio sendromları gibi) da kornea bulanıklığına yol açabileceğinden, sistemik bulgularla birlikte değerlendirilmelidir8).
QMaküler kornea distrofisi nasıl teşhis edilir?
A
Yarık lamba mikroskobunda yaygın buzlu cam benzeri kornea bulanıklığı ve maküler birikintiler görülür; bilateral ve ilerleyici olması, aile öyküsü ve başlangıç yaşı (10-30 yaş) ile klinik tanı konur. Kesin tanı için CHST6 gen testi faydalıdır.
MCD tedavisinin amaçları şunlardır: (1) Görme fonksiyonunun korunması ve iyileştirilmesi, (2) Oküler yüzeyin stabilize edilmesiyle ağrı ve irritasyon semptomlarının hafifletilmesi, (3) Komplikasyonların (epitelyal erozyon, enfeksiyon) önlenmesi. Nedene yönelik bir tedavi olmadığından, hastalık evresi ve semptomlara göre aşamalı müdahaleler yapılır.
Görme azalmasının ilerlediği olgularda kornea nakli tek küratif tedavidir. Endotel tutulumunun varlığına göre cerrahi yöntem seçilir.
Derin anterior lameller keratoplasti (DALK): Endoteli etkilemeyen olgularda ilk seçenektir8,9). Hastanın kendi kornea endotelini koruduğu için greft reddi riski tam kat keratoplastiye göre daha düşüktür. Amerikan Oftalmoloji Akademisi (AAO) değerlendirme raporunda, DALK’ın stromal distrofilerde PKP’ye eşdeğer görme iyileşmesi sağladığı ve daha az endotel kaybına yol açtığı belirtilmiştir8). DALK sonrası nüks oranı düşük olarak bildirilmiştir ve alıcı stromasının değişmesi nedeniyle nüks nadirdir.
Penetran keratoplasti (PKP): Endotel ve Descemet membranında anormal madde birikimi olan ilerlemiş olgular ve santral korneanın ileri derecede inceldiği olgularda endikedir7). MCD için ilk PKP’nin ortalama yaşı 30-40 olarak bildirilmiştir7) ve greft sağkalımı iyidir.
Fototerapötik keratektomi (PTK): Tekrarlayan kornea epitel erozyonları ve yüzeysel skar opasiteleri için semptomatik olarak uygulanır. Ancak hipermetropiye kayma ve stromal opasite indüksiyonuna dikkat edilmelidir.
MCD’de anormal maddeler endotel hücrelerinde de birikebileceğinden, hastalığın derin tabakalara yayıldığı durumlarda DALK yerine PKP tercih edilme eğilimi vardır7). Endotel anormalliği eşlik ediyorsa tam kat keratoplasti endikedir. Ameliyat öncesi ayrıntılı endotel değerlendirmesi (speküler ve konfokal mikroskopi) cerrahi yöntemin belirlenmesinde anahtardır.
DALK sonrası nüks oranı düşük olarak bildirilmiştir ve yaklaşık %5 civarındadır. PKP sonrası greft sağkalımı iyidir ve birçok raporda uzun dönem sağkalım elde edilirken, ameliyattan yıllar sonra anormal materyalin greftte yeniden biriktiği vakalar da bildirilmiştir8). Al-Swailem ve arkadaşlarının Suudi Arabistan serisinde, MCD için PKP sonrası greft sağkalımı iyiydi ancak bazı vakalarda uzun dönemde nüks gözlenmiştir8). Ayrıca, Reinhart ve arkadaşlarının AAO derlemesinde, DALK’ın stromal distrofilerde PKP’ye eşdeğer veya daha iyi görme iyileşmesi ve düşük endotel kaybı oranı sağladığı gösterilmiştir9). Unal ve arkadaşlarının çok merkezli çalışması da MCD dahil stromal distrofilerde DALK’ın etkinliğini bildirmiştir.
Genel olarak, kornea greftinde postoperatif nüks, hastalığın moleküler patofizyolojisinin konakta kalması nedeniyle oluşur. DALK’da konak endoteli ve Descemet membranının ön tabakası korunduğu için, endotel hastalığı olan vakalarda hastalığın ilerleyebileceği unutulmamalıdır. Ameliyat sonrası uzun dönemli düzenli takip (görme keskinliği, yarık lamba muayenesi, endotel hücre yoğunluğu ölçümü) gereklidir.
QDALK mı yoksa PKP mi tercih edilmelidir?
A
Endotel ve Descemet membranı etkilenmemişse derin ön lameller keratoplasti (DALK) ilk seçenektir. DALK, hastanın kendi kornea endotelini koruduğu için reddetme riski daha düşüktür ve postoperatif nüks oranı yaklaşık %5 olarak bildirilmiştir. Öte yandan, endotelde de anormal materyal birikimi olan vakalarda tam kat keratoplasti (PKP) endikedir. Cerrahi yöntem preoperatif endotel değerlendirmesine göre belirlenir.
CHST6 geni, karbonhidrat sülfotransferaz 6 enzimini kodlar3). Bu enzim, keratan molekülündeki N-asetilglukozamine sülfat grubu transferinden sorumludur ve kornea proteoglikanlarında bulunan keratan sülfatın (KS) normal sentezi için gereklidir.
Gen mutasyonu nedeniyle enzim aktivitesinin kaybı, yetersiz sülfatlanmış (hiposülfatlanmış) keratan sülfat sentezine yol açar. Bu anormal keratan sülfat düşük çözünürlüğe sahiptir ve kornea stromasındaki keratositlerin hücre içi ve dışında anormal birikime neden olur2,3).
Keratan sülfatın kantitatif ve kalitatif anormallikleri, aşağıdaki zincirleme patolojik değişikliklere yol açar.
Küçük proteoglikan (SLRP) üretim anormalliği: Lumikan, keratokan ve mimekan gibi korneaya özgü SLRP’ler normal şekilde sentezlenemez.
Kollajen liflerinin düzen anormalliği: Bu SLRP’ler, kornea kollajen liflerinin çapını ve lifler arası mesafeyi sıkı bir şekilde kontrol ederek şeffaflığı sağlar. SLRP fonksiyonundaki azalma, kollajen lif çaplarının heterojen hale gelmesine ve lifler arası mesafenin değişmesine neden olur2).
Hücre dışı matrikste anormal birikim: Sülfatlanmamış keratan sülfatın kendisi hücre dışı matrikste birikir.
Işık saçılımında artış ve şeffaflık kaybı: Yukarıdaki kombine değişiklikler, görünür ışığın saçılımını artırarak korneanın yaygın şekilde bulanıklaşmasına neden olur.
Glikozaminoglikan birikimi, stromal keratositlerin hücre içi ve dışında gözlenir ve hastalık ilerledikçe Bowman membranı, Descemet membranı ve endotel hücrelerine yayılır. Tip I’de, kulak kepçesi kıkırdağında da enzim aktivitesinde azalma doğrulanmıştır ve bu, sistemik bir keratan sülfat metabolizma bozukluğunun parsiyel bir formu olabileceğini düşündürmektedir2). Bununla birlikte, klinik olarak sistemik semptomlar nadirdir ve hastalık, kornea ana semptomu olan lokalize bir bozukluk olarak ele alınır.
Kornea proteoglikanlarından lumikan, kollajen lif çapını yaklaşık 25 nm’de kontrol etme rolünü üstlenirken, keratokan ve mimekan lifler arası mesafeyi düzgün tutar. Bu SLRP’lerin sülfatlanmış yan zincirleri kısa ve eksik olduğunda, kollajen liflerinin kalınlığında heterojenlik oluşur ve lifler arası mesafe de düzensizleşir. Sonuç olarak, kornea stromasında ışık saçılımı artar ve klinik olarak buzlu cam benzeri bir bulanıklık olarak gözlenir.
Kornea şeffaflığının korunması için, Maurer’in kafes teorisi klasik olarak bilinir: “Kollajen liflerinin düzenli kafes benzeri dizilimi, ışık girişimini iptal eder”. Maküler kornea distrofisinde (MCD), bu kafes yapısı SLRP anormalliği nedeniyle bozulur ve şeffaflık kaybolur2,4).
Son temel araştırmalar, CHST6 mutasyonuna bağlı otofaji işlev bozukluğunun keratositlerde piroptozu (inflamatuar hücre ölümü) indükleyerek hastalığın ilerlemesine katkıda bulunabileceğini bildirmiştir8). Benzer otofaji anormallikleri diğer kornea distrofilerinde (granüler tip II gibi) de rapor edilmiştir ve kornea distrofilerinin ortak patogenezi olarak dikkat çekmektedir.
Kalıcı bir tedavi stratejisi olarak gen hedefli tedavi önerilmiştir 8). Meesmann kornea epitel distrofisinde CRISPR/Cas9 ile gen düzenleme üzerine temel araştırmalar ilerlemektedir ve MCD için de gelecekte bir tedavi seçeneği olabilir. Ancak, normal alelin istenmeyen düzenlenmesi (hedef dışı etkiler), kornea stromal hücrelerine etkili gen aktarım yöntemlerinin geliştirilmesi ve uzun dönem güvenliğin doğrulanması gibi klinik uygulama için birçok zorluk devam etmektedir.
Korneada biriken düşük sülfatlı keratan sülfatın enzimatik olarak uzaklaştırılması yaklaşımı temel düzeyde incelenmiştir. Sistemik mukopolisakkaridozda öncü olan enzim replasman tedavisi konseptini korneaya lokal olarak uygulayan bu yöntemin henüz klinik uygulamaya ulaştığına dair bir rapor bulunmamaktadır.
Descemet membran soyulmalı kornea endotel nakli (DSAEK) ve Descemet membran kornea endotel nakli (DMEK) esas olarak Fuchs kornea endotel distrofisi ve büllöz keratopati için geliştirilmiş cerrahi yöntemlerdir, ancak MCD’nin endotel tutulumu ön planda olan vakalarında uygulanmaları da gelecekteki araştırma konuları arasındadır. Şu anda, hem stroma hem de endotelin etkilendiği bu hastalıkta penetran keratoplasti (PKP) pratik seçenektir. Gelecekte, sadece stromayı değiştiren derin anterior lameller keratoplasti (DALK) ile sadece endoteli değiştiren DMEK’in aşamalı olarak kombine edildiği “sıralı keratoplasti” konsepti önerilmiştir, ancak bunların hepsi araştırma aşamasındadır.
Serum keratan sülfat konsantrasyonu, immünofenotip tip II MCD hastalarında normalden düşük olduğu bildirilmiştir ve sistemik bir metabolik belirteç olarak kullanışlılığı tartışılmaktadır 2). Gelecekte, kan testi ile tarama ve CHST6 mutasyonu taşıyan ailelerde erken taşıyıcı tanısı için uygulanması beklenmektedir. Ayrıca, yapay zeka ile yarık lamba görüntülerinin otomatik analizi de bu gibi nadir hastalıkların erken teşhisine katkıda bulunabilecek bir araştırma alanıdır.
Otozomal resesif kalıtım nedeniyle, hastanın kardeşlerinin yaklaşık %25’inde hastalık görülebilir. CHST6 gen testi kullanılarak aile içi tarama ve evlilik/gebelikle ilgili genetik danışmanlık mümkündür. Akraba evliliği öyküsü olan ailelerde veya ailede benzer kornea bulanıklığı olan vakalar varsa, erken dönemde uzmana yönlendirme önerilir.
Akama TO, Nishida K, Nakayama J, et al. Macular corneal dystrophy type I and type II are caused by distinct mutations in a new sulphotransferase gene. Nature Genetics. 2000;26(2):237-241. doi:10.1038/79987.
Aggarwal S, Peck T, Golen J, Karcioglu ZA. Macular corneal dystrophy: A review. Survey of Ophthalmology. 2018;63(5):609-617. doi:10.1016/j.survophthal.2018.03.004.
Jonasson F, Oshima E, Thonar EJ, et al. Macular corneal dystrophy in Iceland. A clinical, genealogic, and immunohistochemical study of 28 patients. Ophthalmology. 1996;103(7):1111-1117. doi:10.1016/s0161-6420(96)30559-9.
Jonasson F, Johannsson JH, Garner A, Rice NS. Macular corneal dystrophy in Iceland. Eye (London, England). 1989;3 ( Pt 4):446-54. doi:10.1038/eye.1989.66. PMID:2606219.
Sultana A, Sridhar MS, Jagannathan A, et al. Novel mutations of the carbohydrate sulfotransferase-6 (CHST6) gene causing macular corneal dystrophy in India. Mol Vis. 2003;9:730-734. PMID:14735064.
Al-Swailem SA, Al-Rajhi AA, Wagoner MD. Penetrating keratoplasty for macular corneal dystrophy. Ophthalmology. 2005;112(2):220-224. doi:10.1016/j.ophtha.2004.08.017.
Reinhart WJ, Musch DC, Jacobs DS, Lee WB, Kaufman SC, Shtein RM. Deep anterior lamellar keratoplasty as an alternative to penetrating keratoplasty a report by the american academy of ophthalmology. Ophthalmology. 2011;118(1):209-218. doi:10.1016/j.ophtha.2010.11.002. PMID:21199711.
Makale metnini kopyalayıp tercih ettiğiniz yapay zeka asistanına yapıştırabilirsiniz.
Makale panoya kopyalandı
Aşağıdaki yapay zeka asistanlarından birini açın ve kopyalanan metni sohbet kutusuna yapıştırın.