有病率の地域差
米国:25万人あたり約0.3人と稀である2,3)
アイスランド:25万人あたり約19人と世界で最も高頻度な地域の一つ5,6)
高有病率地域:南インド・サウジアラビア・アイスランド・北欧で頻度が高い5,7)
その他地域:比較的稀。近親婚や複合ヘテロ接合体で発症する

斑状角膜ジストロフィ(macular corneal dystrophy:MCD)は、角膜実質にグリコサミノグリカン(主に硫酸ケラタン)が蓄積する遺伝性の角膜ジストロフィである。常染色体劣性遺伝形式をとり、第16染色体長腕(16q22)に位置するCHST6遺伝子の変異が原因である1,3)。かつてはGroenouw角膜ジストロフィII型、あるいはフェール角膜ジストロフィとも呼ばれた。
他の多くの角膜実質ジストロフィ(顆粒状・格子状)が常染色体優性遺伝であるのに対し、本疾患は 常染色体劣性遺伝 をとる点が特徴的である。日本では顆粒状角膜ジストロフィ(I型・II型)、格子状角膜ジストロフィ(I型・IIIA型)、膠様滴状角膜ジストロフィとともに四大角膜ジストロフィに数えられ、これらで角膜ジストロフィ全体の約96%を占める。このうち前二者は常染色体優性遺伝、後二者(膠様滴状・斑状)は常染色体劣性遺伝である。
IC3D(International Committee for Classification of Corneal Dystrophies)分類では、MCDは実質ジストロフィの一型として位置付けられる1)。AggarwalらによるSurvey of Ophthalmology 2018年の総説では、本疾患を「稀だが視機能への影響が大きい実質ジストロフィ」として詳述し、診断・治療体系が総括されている4)。IC3D分類第2版では、角膜ジストロフィはカテゴリ1〜4に分類され、原因遺伝子・病理学的所見・臨床像のエビデンス強度により区分される1)。MCDはCHST6遺伝子変異の同定により カテゴリ1(遺伝子レベルで確立されたジストロフィ)に分類されている。
本疾患の歴史的背景としては、1890年にGroenouwによって最初の記載が行われ、後に顆粒状ジストロフィを「I型」、斑状(macular)ジストロフィを「II型」と呼称する流儀が生まれた。1938年にJonesとZimmermanが独立疾患として確立し、2000年のAkamaらによるCHST6遺伝子の同定により分子基盤が明らかとなった3)。
世界的にも地域差が大きく、有病率の高い地域では家族内集積を認める。相対的に稀な疾患である。
有病率の地域差
米国:25万人あたり約0.3人と稀である2,3)
アイスランド:25万人あたり約19人と世界で最も高頻度な地域の一つ5,6)
高有病率地域:南インド・サウジアラビア・アイスランド・北欧で頻度が高い5,7)
その他地域:比較的稀。近親婚や複合ヘテロ接合体で発症する
免疫表現型
I型:角膜・血清ともに硫酸ケラタン陰性2)
IA型:角膜ケラトサイト内に陽性、血清陰性2)
II型:角膜・血清ともに硫酸ケラタン陽性2)
臨床像:3型とも表現型は同一で細隙灯では区別できない2,8)
MCDの免疫表現型は、抗ケラタン硫酸モノクローナル抗体を用いた角膜および血清中の硫酸ケラタン量で分類される2,3)。
| 表現型 | 角膜ケラタン硫酸 | 血清ケラタン硫酸 |
|---|---|---|
| I 型 | 陰性 | 陰性 |
| IA 型 | 陽性(細胞内) | 陰性 |
| II 型 | 陽性 | 陽性 |
ほとんどの患者は I 型または IA 型に分類される。しかし臨床的にはこれら亜型の区別は重要でなく、診察所見では鑑別できない2,8)。
最大の違いは常染色体劣性遺伝をとる点である。顆粒状や格子状角膜ジストロフィは常染色体優性遺伝だが、本疾患はCHST6遺伝子の両アレルに変異が必要である。またびまん性のスリガラス様混濁を呈し、角膜全体が白濁する点、スリット光で見ると中央部は浅層に、周辺部は深層に沈着がある点も特徴的である。
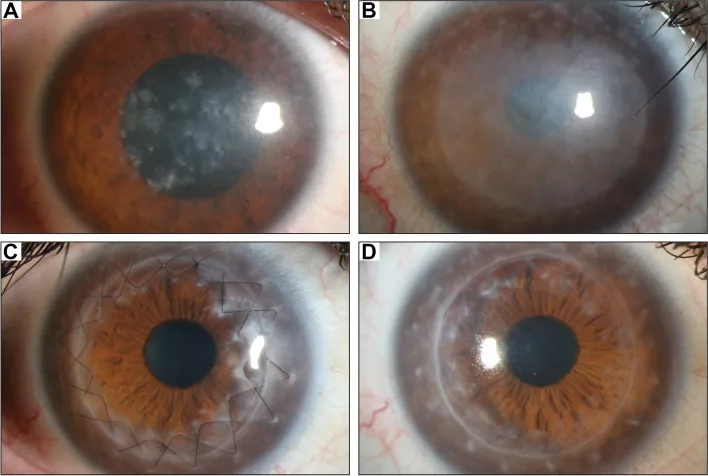
臨床所見の典型的な進行パターンは次の通りである。
臨床的には角膜実質に細かい沈着がびまん性にみられスリガラス様に混濁する。その混濁は進行するにつれて実質全層に及び、また混濁は中央部から周辺に広がる。その後、淡い混濁に加えて、灰白色の小さい不規則な形の多数の混濁が実質浅層〜実質深層にみられる。9)
初期所見
斑点状混濁:角膜中央部の実質浅層に灰白色の小さな斑点状混濁が出現する
スリガラス様混濁:角膜実質にびまん性の淡い混濁を認める
境界不明瞭:混濁の辺縁は不鮮明で、正常実質との境界が不明瞭である
進行期所見
全層への進展:混濁は実質全層に及ぶ
周辺部への拡大:中央から周辺部へ混濁が広がる
角膜菲薄化:中央角膜の厚みが減少する
内皮・Descemet膜への沈着:深部構造にも異常物質が蓄積する
細隙灯顕微鏡では、角膜全体がびまん性に混濁しており、その中に灰白色の不規則な斑状沈着を認める。スリット光で光学切片を作ると、中央部では沈着が浅層に位置し、周辺部では深層に位置する という特徴的な分布を示す。斑状の病変は同心円状に出現するパターンが多い8)。
輪部にも混濁が及ぶことがあり、この点は他の角膜ジストロフィとの重要な鑑別点となる。顆粒状角膜ジストロフィや格子状角膜ジストロフィでは輪部が透明に残ることが多いのに対し、MCDでは角膜全体が輪部まで混濁することが少なくない2,8)。また、不正乱視の出現は前部実質の沈着に関連し、角膜知覚の低下を認めることもある。内皮にも異常物質が沈着するため、進行例では内皮機能低下による実質浮腫を併発することがある8)。
自然経過は個人差があるが、次のような段階をたどることが多い。
MCDは進行性の疾患であり、一生を通じて視機能の低下が続く点で、視力障害が軽度にとどまる顆粒状角膜ジストロフィ I 型や格子状角膜ジストロフィの一部亜型とは予後が大きく異なる4,8)。
原因遺伝子は CHST6(carbohydrate sulfotransferase 6) である3)。16q22に位置し、角膜プロテオグリカン上の N-アセチルグルコサミンへ硫酸基を転移する酵素をコードする。本遺伝子の変異の種類は非常に多く、ミスセンス変異、ナンセンス変異、フレームシフト変異、5’ 上流域の欠失などが各民族で報告されている3,7)。
常染色体劣性遺伝であるため、通常は発端者の両親がともに保因者である。近親婚の多い地域や集団内でとくに発症頻度が高くなる傾向がある。異なる家系同士の結婚で生じる複合ヘテロ接合体でも発症しうる7)。
Akamaらは2000年にCHST6を本疾患の原因遺伝子として同定し、免疫表現型 I 型・II 型がともに同じ遺伝子座の変異によって生じることを示した3)。この発見は、免疫表現型の違いが単一遺伝子の異なる変異パターンによって規定されることを示唆する重要な知見であり、以降の遺伝子診断体制の基礎となった。
報告されている変異の種類は200以上にのぼり、ミスセンス変異が最多である。Sultanaらは南インドの患者群で多数の新規変異を同定し、同地域での高頻度が地域集積的な変異の積み重ねによるものであることを示した7)。サウジアラビアやアイスランドでも類似の地域性集積が報告されており、人口集団の歴史的背景(創始者効果や近親婚の慣習)が発症頻度に寄与していると考えられる5,6,7)。
発症頻度は地域によって異なり、比較的稀な疾患である9)。顆粒状角膜ジストロフィなど他の角膜ジストロフィと比較して発症例が少なく、複合ヘテロ接合体や近親婚背景を持つ家系から報告される傾向にある。
確定診断にはCHST6遺伝子の検査が有用である。遺伝子検査は施設認定を受けた医療機関で実施可能である。ただし多くの施設では細隙灯顕微鏡による臨床診断が主体となる。将来の子どもの発症リスク評価や、臨床所見が典型的でない症例の確定診断に有用である。
MCDを疑うきっかけは「両眼性・進行性・角膜全体がびまん性に混濁する思春期〜若年成人」である。まず自覚症状(視力低下・羞明・刺激感)の経過、家族歴、近親婚歴を詳細に聴取する。次に細隙灯顕微鏡による角膜所見の評価、内皮機能評価、必要に応じて遺伝子検査を行う流れが標準である。
診断の基本となる検査である。充血や角膜浮腫のない両眼性の角膜混濁をみたら角膜ジストロフィを疑う。MCDでは以下の所見が特徴的である。
多くの角膜ジストロフィがスリットレベルで不連続な(沈着間に透明部分がある)病変として観察されるのに対し、MCDでは例外的に びまん性の混濁パターン を呈する。格子状角膜ジストロフィ I 型および膠様滴状角膜ジストロフィとともに「びまん性に観察される角膜沈着」の代表例として特徴付けられる。
確定診断にはCHST6遺伝子の解析が有用である。施設認定を受けた医療機関で実施可能である。
組織学的にはアルシアンブルー染色およびコロイド鉄染色で陽性を示し、角膜実質のケラトサイト細胞内外に低硫酸化グリコサミノグリカンがびまん性に蓄積している像が観察される2,8)。Bowman膜の断裂を認めることがあり、進行例では内皮細胞内にも異常物質が認められる。Descemet膜上に滴状角膜(guttae)様の所見を呈することもある。
| 疾患 | 遺伝形式 | 特徴 |
|---|---|---|
| 顆粒状角膜ジストロフィ | 常染色体優性 | 境界明瞭な顆粒状沈着(間に透明部分あり) |
| 格子状角膜ジストロフィ I 型 | 常染色体優性 | 線状・網目状の格子線(アミロイド) |
| 膠様滴状角膜ジストロフィ | 常染色体劣性 | 桑の実様・帯状の隆起性病変 |
| Schnyder 結晶状角膜ジストロフィ | 常染色体優性 | 針状結晶・高脂血症合併 |
| 先天性角膜実質ジストロフィ | 常染色体優性 | 出生時から存在・実質肥厚 |
| 全身性ムコ多糖症(角膜型) | 劣性/X連鎖 | 全身症状を伴う |
この他、後部不定形角膜ジストロフィ(PACD)、Descemet膜前角膜ジストロフィ(PDCD)も鑑別に挙がる。全身性ムコ多糖症(Hurler、Scheie、Morquio症候群など)も角膜混濁を呈しうるため、全身所見を含めて評価する必要がある8)。
細隙灯顕微鏡でびまん性のスリガラス様角膜混濁と斑状沈着を確認し、両眼性・進行性であること、家族歴、発症年齢(10〜30歳)から臨床診断を行う。確定診断にはCHST6遺伝子検査が有用である。
MCDに対する治療の目的は、(1) 視機能の維持・回復、(2) 眼表面の安定化による疼痛・刺激症状の緩和、(3) 合併症(上皮びらん・感染)の予防の3点に集約される。根本的な病因治療は存在しないため、病期と症状に応じた段階的な介入が行われる。
本疾患の進行を抑制する薬物療法は確立されていない8)。症状緩和を目的に以下を行う。
視力低下が進行した症例では角膜移植術が唯一の根治的手段である。内皮病変の有無により術式を使い分ける。
MCDでは内皮細胞にも異常物質が沈着しうるため、深部まで病変が及んでいる場合はDALKよりPKPが選択される傾向 がある7)。内皮異常を伴う場合は全層角膜移植が適応となる。術前の詳細な内皮評価(スペキュラ・共焦点顕微鏡)が術式決定の要となる。
DALK後の再発率は低く報告されており、約5%程度 とされている。PKP後の移植片生着率は良好で、多くの報告で長期生着が得られる一方、術後数年から十数年の経過で異常物質が移植片に再沈着する例も報告されている8)。Al-Swailemらのサウジアラビアでのシリーズでは、MCDに対するPKP後の生着率は良好だが、一部症例で長期経過中の再発が観察されている8)。また、ReinhartらのAAOレビューでは、実質ジストロフィに対してDALKがPKPと同等以上の視機能回復と低い内皮喪失率を両立することが示されている9)。Unalらの多施設研究でも、MCDを含む実質ジストロフィに対するDALKの有効性が報告されている。
一般に角膜移植片の術後再発は、原疾患の分子病態が宿主側に残存するために生じる。DALKでは宿主の内皮とDescemet膜前層が温存されるため、内皮病変のある症例では病変が進行しうる点に留意する。術後は長期的な定期観察(視力、細隙灯、内皮細胞密度測定)が必要である。
CHST6遺伝子は 炭水化物硫酸転移酵素6(carbohydrate sulfotransferase 6) をコードする3)。この酵素はケラタン分子上のN-アセチルグルコサミンへの硫酸基転移を担い、角膜プロテオグリカンに含まれる硫酸ケラタン(keratan sulfate:KS)の正常な合成に必須である。
遺伝子変異によって酵素活性が失われると、硫酸化が不十分な低硫酸化ケラタン硫酸が合成される。この異常ケラタン硫酸は可溶性が低く、角膜実質のケラトサイトの細胞内外に異常沈着する2,3)。
硫酸ケラタンの量的・質的異常は、以下の連鎖的な病態をもたらす。
グリコサミノグリカンの蓄積は実質ケラトサイトの細胞内外に観察され、病変が進行するとBowman膜・Descemet膜・内皮細胞にも広がる。I 型では耳介軟骨でも酵素活性の低下が確認されており、全身的な硫酸ケラタン代謝異常の部分症である可能性が示唆されている2)。ただし臨床的に全身症状を呈することは乏しく、角膜を主症状とする限局性疾患として扱われる。
角膜プロテオグリカンのうち、ルミカンはコラーゲン線維の直径を約25 nmに制御する役割を担い、ケラトカンとミメカンは線維間隔を均一に保つ。これらSLRPの硫酸化側鎖が短く不完全な状態では、コラーゲン線維は太さにばらつきが生じ、線維間隔も不均一となる。結果として角膜実質内での光散乱が増加し、臨床的にスリガラス様の混濁として観察される。
角膜の透明性維持には「コラーゲン線維の整然とした格子状配列による光干渉の相殺」というMaurerの格子理論が古典的に知られるが、MCDではこの格子構造がSLRP異常により破綻することで透明性が失われる2,4)。
近年の基礎研究では、CHST6変異によるオートファジー(自食作用)の機能障害がケラトサイトのパイロトーシス(炎症性細胞死)を誘導し、疾患進行に寄与する可能性が報告されている8)。他の角膜ジストロフィ(顆粒状II型など)でも同様のオートファジー異常が報告されており、角膜ジストロフィ全般の共通病態として注目されている。
恒久的な治療戦略として遺伝子標的療法が提案されている8)。Meesmann角膜上皮ジストロフィで先行的にCRISPR/Cas9を用いた遺伝子編集の基礎研究が進んでおり、MCDにおいても将来的な治療選択肢となる可能性がある。ただし、正常アレルへの意図しない編集(オフターゲット効果)、角膜実質細胞への効率的な遺伝子導入法の確立、長期安全性の検証など臨床応用には多くの課題が残されている。
角膜内に蓄積した低硫酸化ケラタン硫酸を酵素的に除去するアプローチが基礎的に検討されている。全身性ムコ多糖症で先行している酵素補充療法の概念を角膜局所に応用するものだが、現時点で臨床応用に至った報告はない。
Descemet膜剥離角膜内皮移植術(DSAEK)やDescemet膜角膜内皮移植術(DMEK)は主にFuchs角膜内皮ジストロフィや水疱性角膜症を対象に発展してきた術式だが、MCDの内皮病変先行例に対する応用も今後の研究課題とされる。現時点では実質と内皮の両方が障害される本疾患ではPKPが現実的選択である。将来的には、実質成分のみを置換するDALKと内皮のみを置換するDMEKを段階的に併用する「sequential keratoplasty」の概念も提案されているが、いずれも研究段階である。
血清中の硫酸ケラタン濃度は、免疫表現型 II 型のMCD患者では正常より低値を示すとされ、全身的な代謝マーカーとしての有用性が議論されている2)。将来的には血液検査によるスクリーニングや、CHST6変異家系の早期キャリア診断への応用が期待される。さらに人工知能による細隙灯画像の自動解析も、本疾患のような希少疾患の早期発見に寄与しうる研究領域である。
常染色体劣性遺伝であるため、患者の同胞にはおよそ25%の確率で発症例が含まれうる。CHST6遺伝子検査の利用により、家族内スクリーニングや結婚・妊娠に関する遺伝カウンセリングが可能である。近親婚歴のある家系や、家族内に同様の角膜混濁を有する症例がいる場合には、早期の専門医紹介が推奨される。