Loạn dưỡng giác mạc dạng đốm (MCD) là một loạn dưỡng giác mạc di truyền, trong đó glycosaminoglycan (chủ yếu là keratan sulfat) tích tụ trong nhu mô giác mạc. Bệnh tuân theo kiểu di truyền lặn nhiễm sắc thể thường và do đột biến gen CHST6 nằm trên nhánh dài nhiễm sắc thể 16 (16q22) gây ra1,3). Trước đây còn được gọi là loạn dưỡng giác mạc Groenouw type II hoặc loạn dưỡng giác mạc Fehr.
Khác với nhiều loạn dưỡng nhu mô giác mạc khác (dạng hạt, dạng lưới) có tính di truyền trội nhiễm sắc thể thường, bệnh này có đặc điểm là di truyền lặn nhiễm sắc thể thường. Ở Nhật Bản, bệnh được coi là một trong bốn loại loạn dưỡng giác mạc chính cùng với loạn dưỡng dạng hạt (type I và II), loạn dưỡng dạng lưới (type I và IIIA) và loạn dưỡng dạng giọt keo, chiếm khoảng 96% tổng số các loạn dưỡng giác mạc. Hai loại đầu là di truyền trội nhiễm sắc thể thường, hai loại sau (dạng giọt keo và dạng đốm) là di truyền lặn nhiễm sắc thể thường.
Trong phân loại IC3D (Ủy ban Quốc tế về Phân loại Loạn dưỡng Giác mạc), MCD được xếp là một loại loạn dưỡng nhu mô 1). Trong bài tổng quan của Aggarwal và cộng sự trên Survey of Ophthalmology năm 2018, bệnh này được mô tả là “loạn dưỡng nhu mô hiếm gặp nhưng ảnh hưởng lớn đến chức năng thị giác”, và hệ thống chẩn đoán và điều trị được tóm tắt 4). Trong phiên bản thứ 2 của phân loại IC3D, loạn dưỡng giác mạc được phân loại thành các hạng mục 1–4 dựa trên mức độ bằng chứng về gen gây bệnh, kết quả giải phẫu bệnh và hình ảnh lâm sàng 1). MCD được xếp vào hạng mục 1 (loạn dưỡng đã được xác định ở cấp độ gen) nhờ xác định đột biến gen CHST6.
Về mặt lịch sử, bệnh này được mô tả lần đầu bởi Groenouw vào năm 1890, sau đó hình thành truyền thống gọi loạn dưỡng dạng hạt là “type I” và loạn dưỡng dạng đốm là “type II”. Năm 1938, Jones và Zimmerman xác lập nó như một bệnh độc lập, và năm 2000, cơ sở phân tử được làm sáng tỏ bởi Akama và cộng sự thông qua xác định gen CHST6 3).
Trên toàn cầu, có sự khác biệt địa lý lớn, và ở những khu vực có tỷ lệ mắc cao, có sự tập trung trong gia đình. Đây là một bệnh tương đối hiếm.
Khác biệt địa lý về tỷ lệ mắc
Hoa Kỳ: khoảng 0,3 trên 250.000 người, hiếm 2,3)
Iceland: khoảng 19 trên 250.000 người, một trong những khu vực có tần suất cao nhất thế giới 5,6)
Khu vực có tỷ lệ mắc cao: Nam Ấn Độ, Ả Rập Saudi, Iceland và Bắc Âu 5,7)
Khu vực khác: tương đối hiếm. Xảy ra trong hôn nhân cận huyết hoặc dị hợp tử kép
Kiểu hình miễn dịch
Type I: âm tính với keratan sulfate ở giác mạc và huyết thanh 2)
Type IA: dương tính trong tế bào keratocyte giác mạc, âm tính trong huyết thanh 2)
Type II: dương tính với keratan sulfate ở giác mạc và huyết thanh 2)
Hình ảnh lâm sàng: Kiểu hình của cả ba type giống hệt nhau và không thể phân biệt bằng đèn khe 2,8)
Kiểu hình miễn dịch của MCD được phân loại dựa trên lượng keratan sulfate trong giác mạc và huyết thanh sử dụng kháng thể đơn dòng kháng keratan sulfate 2,3).
Kiểu hình
Keratan sulfat giác mạc
Keratan sulfat huyết thanh
Loại I
Âm tính
Âm tính
Loại IA
Dương tính (nội bào)
Âm tính
Loại II
Dương tính
Dương tính
Hầu hết bệnh nhân được phân loại là loại I hoặc IA. Tuy nhiên, về mặt lâm sàng, sự phân biệt giữa các phân nhóm này không quan trọng và không thể phân biệt qua thăm khám2,8).
QLoạn dưỡng giác mạc dạng đốm khác với các loạn dưỡng giác mạc khác như thế nào?
A
Sự khác biệt lớn nhất là di truyền lặn trên nhiễm sắc thể thường. Loạn dưỡng giác mạc dạng hạt và dạng lưới di truyền trội trên nhiễm sắc thể thường, nhưng bệnh này cần đột biến ở cả hai alen của gen CHST6. Ngoài ra, bệnh biểu hiện đục lan tỏa dạng kính mờ, toàn bộ giác mạc trở nên trắng đục, và khi khám bằng đèn khe, có lắng đọng ở lớp nông ở trung tâm và lớp sâu ở ngoại vi.
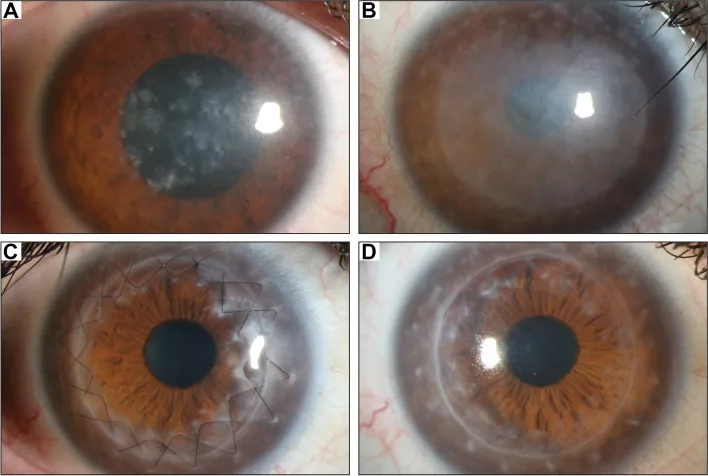
Gassel CJ, et al. Histological findings of corneal tissue after failed phototherapeutic keratectomy in macular corneal dystrophy - a case report. BMC Ophthalmol. 2022. Figure 1. PMCID: PMC9080147. License: CC BY.
Ảnh đèn khe cho thấy độ mờ khuếch tán màu xám trắng và lắng đọng dạng đốm từ trung tâm ra toàn bộ giác mạc. Thể hiện các dấu hiệu lâm sàng điển hình của loạn dưỡng giác mạc dạng đốm, giúp dễ hiểu về độ mờ giác mạc gây giảm thị lực.
Mô hình tiến triển điển hình của các dấu hiệu lâm sàng như sau.
Về mặt lâm sàng, các lắng đọng mịn khuếch tán được thấy trong nhu mô giác mạc, gây ra độ mờ như kính mờ. Khi tiến triển, độ mờ lan rộng ra toàn bộ chiều dày nhu mô và lan từ trung tâm ra ngoại vi. Sau đó, ngoài độ mờ nhạt, nhiều độ mờ nhỏ không đều màu trắng xám được thấy ở nhu mô nông đến sâu.9)
Dấu hiệu ban đầu
Độ mờ dạng chấm: Các độ mờ dạng chấm nhỏ màu trắng xám xuất hiện ở nhu mô nông của giác mạc trung tâm
Độ mờ như kính mờ: Độ mờ nhạt khuếch tán được thấy ở nhu mô giác mạc
Ranh giới không rõ: Các mép của độ mờ không sắc nét, ranh giới với nhu mô bình thường không rõ ràng
Dấu hiệu giai đoạn tiến triển
Lan rộng ra toàn bộ các lớp: Đục lan rộng ra toàn bộ chiều dày nhu mô
Lan rộng ra vùng ngoại vi: Đục lan từ trung tâm ra ngoại vi
Mỏng giác mạc: Độ dày giác mạc trung tâm giảm
Lắng đọng ở nội mô và màng Descemet: Chất bất thường cũng tích tụ ở các cấu trúc sâu
Trên sinh hiển vi đèn khe, toàn bộ giác mạc bị đục lan tỏa với các mảng lắng đọng màu xám trắng không đều. Khi tạo lát cắt quang học bằng chùm sáng khe, thấy sự phân bố đặc trưng: ở trung tâm, lắng đọng nằm ở lớp nông; ở ngoại vi, lắng đọng nằm ở lớp sâu. Các tổn thương dạng mảng thường xuất hiện theo hình tròn đồng tâm8).
Đục có thể lan đến vùng rìa, và đây là điểm phân biệt quan trọng với các loạn dưỡng giác mạc khác. Trong loạn dưỡng giác mạc dạng hạt và loạn dưỡng giác mạc dạng lưới, vùng rìa thường trong suốt, trong khi ở MCD, toàn bộ giác mạc thường bị đục đến tận rìa2,8). Ngoài ra, sự xuất hiện của loạn thị không đều liên quan đến lắng đọng ở nhu mô trước, và có thể giảm cảm giác giác mạc. Do chất bất thường cũng lắng đọng ở nội mô, trong trường hợp tiến triển có thể xảy ra phù nhu mô do suy giảm chức năng nội mô8).
Diễn tiến tự nhiên khác nhau giữa các cá nhân, nhưng thường trải qua các giai đoạn sau.
Trẻ nhỏ (giai đoạn không triệu chứng): Đột biến gen đã có từ khi sinh, nhưng dấu hiệu trên sinh hiển vi khe đèn rất ít và không có triệu chứng
Tuổi đi học đến thiếu niên (giai đoạn đục ban đầu): Đục lan tỏa nhẹ xuất hiện ở lớp nông của nhu mô giác mạc, sau đó thấy lắng đọng dạng mảng
10-30 tuổi (giai đoạn suy giảm thị lực): Đục tiến triển, bệnh nhân bắt đầu nhận thấy giảm thị lực
30-40 tuổi (giai đoạn tiến triển): Đục lan rộng ra toàn bộ nhu mô và vùng rìa, giảm cảm giác giác mạc, mỏng giác mạc và loạn thị không đều trở nên rõ ràng
Trung niên trở lên (giai đoạn chỉ định ghép giác mạc): Suy giảm chức năng thị giác đạt mức ảnh hưởng đến sinh hoạt hàng ngày, và ghép giác mạc được xem xét
Gen gây bệnh là CHST6 (carbohydrate sulfotransferase 6)3). Nằm trên nhiễm sắc thể 16q22, mã hóa enzyme chuyển nhóm sulfat đến N-acetylglucosamine trên proteoglycan giác mạc. Các loại đột biến của gen này rất đa dạng, bao gồm đột biến sai nghĩa, vô nghĩa, dịch khung và mất đoạn ở vùng thượng nguồn 5’, đã được báo cáo ở nhiều nhóm dân tộc khác nhau 3,7).
Vì di truyền là lặn trên nhiễm sắc thể thường, thông thường cả cha và mẹ của người bệnh đều là người mang gen. Tần suất mắc bệnh có xu hướng cao hơn ở các khu vực hoặc nhóm có nhiều hôn nhân cận huyết. Bệnh cũng có thể xảy ra ở thể dị hợp tử kép do kết hôn giữa các gia đình khác nhau 7).
Akama và cộng sự năm 2000 đã xác định CHST6 là gen gây bệnh này và chỉ ra rằng cả hai kiểu hình miễn dịch loại I và loại II đều do đột biến tại cùng một locus gen 3). Phát hiện này rất quan trọng, cho thấy sự khác biệt về kiểu hình miễn dịch được quyết định bởi các dạng đột biến khác nhau trên một gen duy nhất, và đã trở thành nền tảng cho hệ thống chẩn đoán di truyền sau này.
Hơn 200 loại đột biến đã được báo cáo, trong đó đột biến sai nghĩa là phổ biến nhất. Sultana và cộng sự đã xác định nhiều đột biến mới ở một nhóm bệnh nhân Nam Ấn Độ và chỉ ra rằng tần suất cao ở khu vực đó là do sự tích tụ các đột biến đặc thù địa phương 7). Sự tích tụ tương tự cũng được báo cáo ở Ả Rập Saudi và Iceland, và người ta cho rằng bối cảnh lịch sử dân số (hiệu ứng người sáng lập và tập quán hôn nhân cận huyết) góp phần vào tần suất mắc bệnh 5,6,7).
Tần suất mắc bệnh khác nhau theo khu vực, và đây là một bệnh tương đối hiếm 9). So với các loạn dưỡng giác mạc khác như loạn dưỡng giác mạc dạng hạt, số ca bệnh ít hơn và thường được báo cáo ở các gia đình có tiền sử hôn nhân cận huyết hoặc dị hợp tử kép.
Tiền sử gia đình: Vì di truyền lặn trên nhiễm sắc thể thường, cả cha và mẹ đều phải là người mang gen
Hôn nhân cận huyết: Làm tăng tỷ lệ mắc bệnh
Yếu tố địa lý: Tỷ lệ mắc cao ở Nam Ấn Độ, Ả Rập Saudi, Iceland và Scandinavia 5,7)
QXét nghiệm di truyền có cần thiết không?
A
Xét nghiệm gen CHST6 hữu ích cho chẩn đoán xác định. Xét nghiệm di truyền có thể được thực hiện tại các cơ sở y tế được chứng nhận. Tuy nhiên, tại nhiều cơ sở, chẩn đoán lâm sàng chủ yếu dựa trên khám bằng đèn khe. Xét nghiệm di truyền hữu ích để đánh giá nguy cơ cho con cái trong tương lai và để xác nhận chẩn đoán trong các trường hợp có biểu hiện lâm sàng không điển hình.
Nghi ngờ MCD xuất hiện ở thanh thiếu niên đến người trẻ tuổi có đục giác mạc lan tỏa toàn bộ, hai bên và tiến triển. Đầu tiên, hỏi chi tiết về triệu chứng chủ quan (giảm thị lực, sợ ánh sáng, kích ứng), tiền sử gia đình và hôn nhân cận huyết. Sau đó, đánh giá giác mạc bằng đèn khe, đánh giá chức năng nội mô, và nếu cần, xét nghiệm di truyền.
Đây là xét nghiệm cơ bản để chẩn đoán. Nếu thấy đục giác mạc hai bên không có xung huyết hoặc phù nề, nghi ngờ loạn dưỡng giác mạc. Trong MCD, các dấu hiệu sau đây đặc trưng:
Đục lan tỏa dạng kính mờ: Toàn bộ giác mạc bị đục lan tỏa
Lắng đọng dạng đốm: Nhiều đám đục không đều màu xám trắng
Phân bố đồng tâm: Khi cắt bằng chùm sáng khe, lớp nông ở trung tâm và lớp sâu ở ngoại vi
Thâm nhiễm rìa: Đục có thể lan đến rìa giác mạc
Bất thường bề mặt nội mô: Ở giai đoạn tiến triển, có thể thấy lắng đọng dạng giọt
Trong khi hầu hết các loạn dưỡng giác mạc được quan sát ở mức khe như các tổn thương không liên tục (có vùng trong suốt giữa các lắng đọng), MCD đặc biệt có dạng đục lan tỏa. Cùng với loạn dưỡng giác mạc dạng lưới type I và loạn dưỡng giác mạc dạng giọt keo, đây là ví dụ điển hình của “lắng đọng giác mạc được quan sát lan tỏa”.
Chụp cắt lớp quang học kết hợp đoạn trước (AS-OCT): Hình ảnh hóa sự phân bố lắng đọng ở lớp nông và sâu của giác mạc
Kính hiển vi đồng tiêu (in vivo confocal microscopy): Cho thấy vật liệu phản xạ cao với ranh giới không rõ và mất hình ảnh tế bào giác mạc bình thường8)
Kính hiển vi siêu âm sinh học (UBM): Hữu ích để đánh giá độ đục sâu và cấu trúc phía sau giác mạc
Kính hiển vi đặc tả (Specular Microscope): Đánh giá mật độ và hình thái tế bào nội mô. Mức độ tổn thương nội mô liên quan trực tiếp đến lựa chọn phương pháp phẫu thuật
Pentacam giác mạc (Máy chụp Scheimpflug): Cung cấp bản đồ mật độ toàn bộ các lớp giác mạc, hữu ích để đánh giá ba chiều các vùng đục
Về mặt mô học, nhuộm Alcian blue và nhuộm sắt keo cho kết quả dương tính, và quan sát thấy sự tích tụ lan tỏa của glycosaminoglycan sulfat thấp trong và ngoài tế bào keratocyte của nhu mô giác mạc2,8). Có thể thấy đứt màng Bowman, và trong các trường hợp tiến triển, chất bất thường cũng được tìm thấy trong tế bào nội mô. Có thể có các biểu hiện giống như giọt (guttae) trên màng Descemet.
Bệnh mucopolysaccharidosis toàn thân (thể giác mạc)
Lặn/liên kết X
Kèm triệu chứng toàn thân
Ngoài ra, loạn dưỡng giác mạc sau dạng không định hình (PACD) và loạn dưỡng giác mạc trước màng Descemet (PDCD) cũng nằm trong chẩn đoán phân biệt. Bệnh mucopolysaccharidosis toàn thân (như hội chứng Hurler, Scheie, Morquio) cũng có thể gây đục giác mạc, do đó cần đánh giá bao gồm các biểu hiện toàn thân8).
QLoạn dưỡng giác mạc dạng đốm được chẩn đoán như thế nào?
A
Chẩn đoán lâm sàng bằng đèn khe cho thấy đục giác mạc lan tỏa dạng kính mờ và lắng đọng dạng đốm, kèm hai mắt, tiến triển, tiền sử gia đình và tuổi khởi phát (10-30 tuổi). Xét nghiệm di truyền CHST6 hữu ích để xác nhận.
Mục tiêu điều trị MCD là: (1) duy trì hoặc phục hồi chức năng thị giác, (2) giảm đau và triệu chứng kích ứng bằng cách ổn định bề mặt mắt, (3) ngăn ngừa biến chứng (xói mòn biểu mô và nhiễm trùng). Vì không có liệu pháp căn nguyên triệt để, can thiệp được thực hiện theo từng giai đoạn dựa trên giai đoạn bệnh và triệu chứng.
Trong các trường hợp suy giảm thị lực tiến triển, ghép giác mạc là phương pháp điều trị triệt để duy nhất. Kỹ thuật được lựa chọn dựa trên sự hiện diện hay không của bệnh nội mô.
Ghép giác mạc lớp trước sâu (DALK): Là lựa chọn đầu tiên trong các trường hợp không có tổn thương nội mô 8,9). Do bảo tồn nội mô giác mạc của chính bệnh nhân, nguy cơ thải ghép thấp hơn so với ghép xuyên thấu. Trong báo cáo đánh giá của AAO (Học viện Nhãn khoa Hoa Kỳ), DALK cho loạn dưỡng nhu mô được đánh giá là phục hồi chức năng thị giác tương đương PKP với mất tế bào nội mô ít hơn 8). Tỷ lệ tái phát sau DALK thấp và tái phát hiếm gặp vì nhu mô giác mạc của người nhận được thay thế.
Ghép giác mạc xuyên thấu (PKP): Chỉ định trong các trường hợp tiến triển có tổn thương nội mô hoặc màng Descemet với lắng đọng chất bất thường, và các trường hợp giác mạc trung tâm mỏng nghiêm trọng 7). Tuổi trung bình cho PKP đầu tiên do MCD là 30-40 tuổi 7), và tỷ lệ sống của mảnh ghép tốt.
Cắt giác mạc điều trị bằng laser (PTK): Được thực hiện triệu chứng cho xói mòn biểu mô tái phát hoặc đục sẹo nông. Tuy nhiên, cần lưu ý đến tình trạng viễn thị hóa và gây đục nhu mô.
Trong MCD, chất bất thường cũng có thể lắng đọng trong tế bào nội mô, do đó trong các trường hợp bệnh lan rộng đến lớp sâu, PKP có xu hướng được chọn hơn DALK7). Nếu có bất thường nội mô, ghép xuyên thấu được chỉ định. Đánh giá nội mô chi tiết trước phẫu thuật (kính hiển vi đặc tả hoặc đồng tiêu) là chìa khóa để quyết định kỹ thuật phẫu thuật.
Tỷ lệ tái phát sau DALK được báo cáo là thấp, khoảng 5%. Trong khi tỷ lệ sống của mảnh ghép sau PKP tốt, nhiều báo cáo cho thấy sự sống lâu dài, nhưng một số trường hợp ghi nhận sự lắng đọng lại chất bất thường trên mảnh ghép sau vài năm đến hàng chục năm phẫu thuật8). Trong loạt nghiên cứu của Al-Swailem và cộng sự tại Ả Rập Saudi, tỷ lệ sống của mảnh ghép sau PKP cho MCD tốt, nhưng tái phát được quan sát thấy ở một số trường hợp trong quá trình theo dõi dài hạn8). Ngoài ra, trong đánh giá của AAO bởi Reinhart và cộng sự, DALK đã được chứng minh đạt được phục hồi thị lực tương đương hoặc tốt hơn PKP với tỷ lệ mất tế bào nội mô thấp9). Nghiên cứu đa trung tâm của Unal và cộng sự cũng báo cáo hiệu quả của DALK trong các chứng loạn dưỡng nhu mô bao gồm MCD.
Nói chung, tái phát sau ghép giác mạc xảy ra do bệnh lý phân tử của bệnh nguyên phát vẫn còn ở bên vật chủ. Trong DALK, nội mô của vật chủ và lớp trước màng Descemet được bảo tồn, do đó cần lưu ý rằng bệnh có thể tiến triển ở những trường hợp có tổn thương nội mô. Sau phẫu thuật, cần theo dõi định kỳ dài hạn (kiểm tra thị lực, đèn khe, đo mật độ tế bào nội mô).
QNên chọn DALK hay PKP?
A
Nếu nội mô hoặc màng Descemet không bị ảnh hưởng, ghép lớp trước sâu (DALK) là lựa chọn đầu tiên. DALK bảo tồn nội mô giác mạc của chính bệnh nhân, do đó giảm nguy cơ thải ghép, và tỷ lệ tái phát được báo cáo khoảng 5% sau phẫu thuật. Mặt khác, trong những trường hợp có lắng đọng chất bất thường cả ở nội mô, ghép giác mạc toàn bộ độ dày (PKP) được chỉ định. Kỹ thuật phẫu thuật được xác định dựa trên đánh giá nội mô trước phẫu thuật.
Gen CHST6 mã hóa carbohydrate sulfotransferase 63). Enzyme này chịu trách nhiệm chuyển nhóm sulfat đến N-acetylglucosamine trên phân tử keratan, và cần thiết cho quá trình tổng hợp bình thường keratan sulfat (KS) có trong proteoglycan giác mạc.
Khi hoạt động enzyme bị mất do đột biến gen, keratan sulfat sulfat hóa thấp và không hoàn chỉnh được tổng hợp. Keratan sulfat bất thường này có độ hòa tan thấp và lắng đọng bất thường bên trong và bên ngoài tế bào keratocyte trong nhu mô giác mạc2,3).
Các bất thường về số lượng và chất lượng của keratan sulfat dẫn đến chuỗi bệnh lý sau đây.
Sản xuất bất thường proteoglycan nhỏ giàu leucine (SLRP): Các SLRP đặc trưng của giác mạc như lumican, keratocan và mimecan không được tổng hợp bình thường
Bất thường sắp xếp sợi collagen: Các SLRP này kiểm soát chặt chẽ đường kính sợi collagen giác mạc và khoảng cách giữa các sợi, đảm bảo độ trong suốt. Suy giảm chức năng SLRP dẫn đến đường kính sợi collagen không đồng nhất và khoảng cách giữa các sợi thay đổi 2)
Tích tụ bất thường trong chất nền ngoại bào: Bản thân keratan không sulfat hóa lắng đọng trong chất nền ngoại bào
Tăng tán xạ ánh sáng và mất độ trong suốt: Các thay đổi kết hợp trên làm tăng tán xạ ánh sáng khả kiến, gây đục lan tỏa toàn bộ giác mạc
Sự tích tụ glycosaminoglycan được quan sát thấy bên trong và bên ngoài tế bào keratocyte của nhu mô, và khi bệnh tiến triển, lan rộng đến màng Bowman, màng Descemet và tế bào nội mô. Ở type I, sự giảm hoạt động enzyme cũng đã được xác nhận ở sụn vành tai, cho thấy khả năng đây là một phần của rối loạn chuyển hóa keratan sulfat toàn thân 2). Tuy nhiên, các triệu chứng toàn thân hiếm khi biểu hiện lâm sàng, và bệnh được coi là bệnh khu trú với triệu chứng chính ở giác mạc.
Trong số các proteoglycan giác mạc, lumican đóng vai trò kiểm soát đường kính sợi collagen ở khoảng 25 nm, trong khi keratocan và mimecan duy trì khoảng cách đều đặn giữa các sợi. Khi các chuỗi bên sulfat hóa của các SLRP này ngắn và không hoàn chỉnh, các sợi collagen có độ dày không đồng đều và khoảng cách giữa các sợi trở nên không đều. Kết quả là sự tán xạ ánh sáng trong nhu mô giác mạc tăng lên, và trên lâm sàng được quan sát thấy dưới dạng đục như kính mờ.
Lý thuyết mạng lưới Maurer được biết đến kinh điển để duy trì độ trong suốt của giác mạc thông qua “sự triệt tiêu giao thoa ánh sáng nhờ sự sắp xếp mạng lưới có trật tự của các sợi collagen”, nhưng trong MCD, cấu trúc mạng lưới này bị phá vỡ do bất thường SLRP, dẫn đến mất độ trong suốt 2,4).
Trong nghiên cứu cơ bản gần đây, đã có báo cáo rằng rối loạn chức năng autophagy (tự thực) do đột biến CHST6 có thể gây ra pyroptosis (chết tế bào viêm) của keratocyte, góp phần vào sự tiến triển của bệnh 8). Các bất thường autophagy tương tự cũng đã được báo cáo trong các chứng loạn dưỡng giác mạc khác (như type hạt II), thu hút sự chú ý như một cơ chế bệnh lý chung của loạn dưỡng giác mạc nói chung.
Liệu pháp gen nhắm mục tiêu đã được đề xuất như một chiến lược điều trị vĩnh viễn 8). Nghiên cứu cơ bản về chỉnh sửa gen bằng CRISPR/Cas9 đang được tiến hành trên bệnh loạn dưỡng biểu mô giác mạc Meesmann và có thể trở thành lựa chọn điều trị trong tương lai cho MCD. Tuy nhiên, vẫn còn nhiều thách thức cho ứng dụng lâm sàng, như chỉnh sửa không chủ ý các alen bình thường (hiệu ứng ngoài mục tiêu), phát triển các phương pháp đưa gen hiệu quả vào tế bào mô đệm giác mạc và xác minh tính an toàn lâu dài.
Một cách tiếp cận để loại bỏ keratan sulfate sulfat hóa thấp tích tụ trong giác mạc bằng enzyme đang được nghiên cứu cơ bản. Điều này áp dụng khái niệm liệu pháp thay thế enzyme được sử dụng trong bệnh mucopolysaccharidosis toàn thân một cách cục bộ vào giác mạc, nhưng hiện chưa có báo cáo về ứng dụng lâm sàng.
Phẫu thuật ghép nội mô giác mạc tự động bóc màng Descemet (DSAEK) và ghép nội mô màng Descemet (DMEK) là các kỹ thuật được phát triển chủ yếu cho bệnh loạn dưỡng nội mô Fuchs và bệnh giác mạc bọng nước, nhưng ứng dụng của chúng trên các trường hợp MCD có tổn thương nội mô sớm là chủ đề nghiên cứu trong tương lai. Hiện tại, vì bệnh ảnh hưởng đến cả mô đệm và nội mô, ghép giác mạc xuyên thấu (PKP) là lựa chọn thực tế. Trong tương lai, khái niệm “ghép giác mạc tuần tự” kết hợp ghép giác mạc lớp trước sâu (DALK) để thay thế mô đệm và DMEK để thay thế nội mô theo từng giai đoạn đã được đề xuất, nhưng tất cả vẫn đang trong giai đoạn nghiên cứu.
Nồng độ keratan sulfate trong huyết thanh được báo cáo là thấp hơn bình thường ở bệnh nhân MCD có kiểu hình miễn dịch loại II, và tính hữu ích của nó như một dấu ấn chuyển hóa toàn thân đang được thảo luận 2). Trong tương lai, nó được kỳ vọng sẽ được sử dụng để sàng lọc qua xét nghiệm máu và chẩn đoán sớm người mang đột biến trong các gia đình có CHST6. Ngoài ra, phân tích tự động hình ảnh đèn khe bằng trí tuệ nhân tạo cũng có thể góp phần phát hiện sớm các bệnh hiếm gặp như thế này.
Vì bệnh di truyền lặn trên nhiễm sắc thể thường, khoảng 25% anh chị em ruột của bệnh nhân có thể bị ảnh hưởng. Bằng cách sử dụng xét nghiệm gen CHST6, có thể thực hiện sàng lọc gia đình và tư vấn di truyền về hôn nhân và mang thai. Nên giới thiệu sớm đến bác sĩ chuyên khoa ở những gia đình có tiền sử hôn nhân cận huyết hoặc có trường hợp đục giác mạc tương tự trong gia đình.
Akama TO, Nishida K, Nakayama J, et al. Macular corneal dystrophy type I and type II are caused by distinct mutations in a new sulphotransferase gene. Nature Genetics. 2000;26(2):237-241. doi:10.1038/79987.
Aggarwal S, Peck T, Golen J, Karcioglu ZA. Macular corneal dystrophy: A review. Survey of Ophthalmology. 2018;63(5):609-617. doi:10.1016/j.survophthal.2018.03.004.
Jonasson F, Oshima E, Thonar EJ, et al. Macular corneal dystrophy in Iceland. A clinical, genealogic, and immunohistochemical study of 28 patients. Ophthalmology. 1996;103(7):1111-1117. doi:10.1016/s0161-6420(96)30559-9.
Jonasson F, Johannsson JH, Garner A, Rice NS. Macular corneal dystrophy in Iceland. Eye (London, England). 1989;3 ( Pt 4):446-54. doi:10.1038/eye.1989.66. PMID:2606219.
Sultana A, Sridhar MS, Jagannathan A, et al. Novel mutations of the carbohydrate sulfotransferase-6 (CHST6) gene causing macular corneal dystrophy in India. Mol Vis. 2003;9:730-734. PMID:14735064.
Al-Swailem SA, Al-Rajhi AA, Wagoner MD. Penetrating keratoplasty for macular corneal dystrophy. Ophthalmology. 2005;112(2):220-224. doi:10.1016/j.ophtha.2004.08.017.
Reinhart WJ, Musch DC, Jacobs DS, Lee WB, Kaufman SC, Shtein RM. Deep anterior lamellar keratoplasty as an alternative to penetrating keratoplasty a report by the american academy of ophthalmology. Ophthalmology. 2011;118(1):209-218. doi:10.1016/j.ophtha.2010.11.002. PMID:21199711.
Sao chép toàn bộ bài viết và dán vào trợ lý AI bạn muốn dùng.
Đã sao chép bài viết vào clipboard
Mở một trợ lý AI bên dưới và dán nội dung đã sao chép vào ô chat.