La distrofia corneal macular (macular corneal dystrophy: MCD) es una distrofia corneal hereditaria en la que se acumulan glucosaminoglicanos (principalmente sulfato de queratán) en el estroma corneal. Sigue un patrón de herencia autosómico recesivo y es causada por mutaciones en el gen CHST6 ubicado en el brazo largo del cromosoma 16 (16q22)1,3). Anteriormente se llamaba distrofia corneal de Groenouw tipo II o distrofia corneal de Fehr.
Mientras que muchas otras distrofias del estroma corneal (granular, reticular) son autosómicas dominantes, esta enfermedad se caracteriza por ser autosómica recesiva. En Japón, se considera una de las cuatro grandes distrofias corneales junto con la distrofia corneal granular (tipos I y II), la distrofia corneal reticular (tipos I y IIIA) y la distrofia corneal gelatinosa en gotas, representando aproximadamente el 96% de todas las distrofias corneales. De estas, las dos primeras son autosómicas dominantes, mientras que las dos últimas (gelatinosa en gotas y macular) son autosómicas recesivas.
En la clasificación IC3D (Comité Internacional para la Clasificación de las Distrofias Corneales), la MCD se ubica como un tipo de distrofia estromal 1). En una revisión de 2018 de Aggarwal et al. en Survey of Ophthalmology, esta enfermedad se detalla como “una distrofia estromal rara pero visualmente significativa”, y se resume el sistema de diagnóstico y tratamiento 4). En la segunda edición de la clasificación IC3D, las distrofias corneales se clasifican en categorías 1 a 4, diferenciadas por la fuerza de la evidencia de genes causantes, hallazgos patológicos y características clínicas 1). La MCD se clasifica como Categoría 1 (una distrofia establecida a nivel genético) debido a la identificación de mutaciones en el gen CHST6.
Históricamente, esta enfermedad fue descrita por primera vez por Groenouw en 1890, y más tarde surgió una convención que llamaba distrofia granular “tipo I” y distrofia macular “tipo II”. En 1938, Jones y Zimmerman la establecieron como una enfermedad independiente, y en 2000, Akama et al. identificaron el gen CHST6, aclarando su base molecular 3).
A nivel mundial, existen grandes diferencias regionales, y en áreas con alta prevalencia se observa agregación familiar. Es una enfermedad relativamente rara.
Diferencias Regionales en la Prevalencia
Estados Unidos: Rara, aproximadamente 0.3 por 250,000 personas 2,3)
Islandia: Aproximadamente 19 por 250,000 personas, una de las frecuencias más altas del mundo 5,6)
Regiones de alta prevalencia: Sur de India, Arabia Saudita, Islandia y el norte de Europa tienen alta frecuencia 5,7)
Otras regiones: Relativamente rara. Ocurre con matrimonios consanguíneos o heterocigosidad compuesta.
Inmunofenotipos
Tipo I: Sulfato de queratán negativo tanto en córnea como en suero 2)
Tipo IA: Positivo en queratocitos corneales, negativo en suero 2)
Tipo II: Sulfato de queratán positivo tanto en córnea como en suero 2)
Características clínicas: Los tres tipos tienen fenotipos idénticos y no se pueden distinguir con lámpara de hendidura2,8)
Los inmunofenotipos de la MCD se clasifican por la cantidad de sulfato de queratán en la córnea y el suero utilizando anticuerpos monoclonales anti-sulfato de queratán 2,3).
Fenotipo
Sulfato de queratán corneal
Sulfato de queratán sérico
Tipo I
Negativo
Negativo
Tipo IA
Positivo (intracelular)
Negativo
Tipo II
Positivo
Positivo
La mayoría de los pacientes se clasifican como tipo I o IA. Sin embargo, clínicamente, distinguir estos subtipos no es importante y no se pueden diferenciar mediante los hallazgos del examen 2,8).
Q¿En qué se diferencia la distrofia corneal macular de otras distrofias corneales?
A
La mayor diferencia es que sigue un patrón de herencia autosómico recesivo. Las distrofias corneales granular y reticular son autosómicas dominantes, pero esta enfermedad requiere mutaciones en ambos alelos del gen CHST6. También se presenta con opacidades difusas en vidrio esmerilado, causando que toda la córnea se vuelva turbia, y en el examen con lámpara de hendidura se observan depósitos en la capa superficial centralmente y en la capa profunda periféricamente.
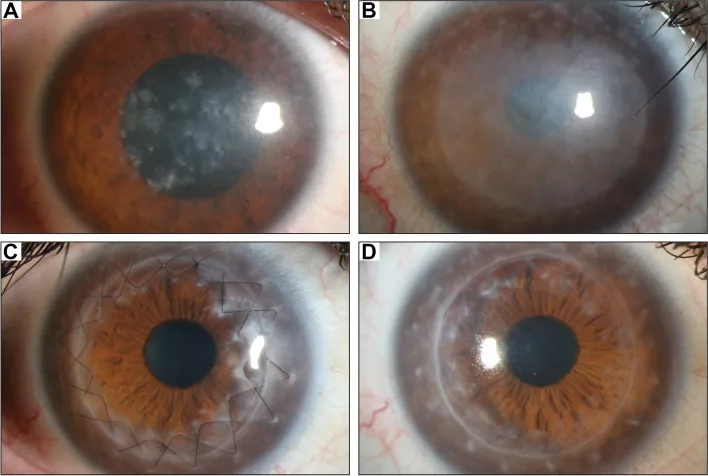
Gassel CJ, et al. Histological findings of corneal tissue after failed phototherapeutic keratectomy in macular corneal dystrophy - a case report. BMC Ophthalmol. 2022. Figure 1. PMCID: PMC9080147. License: CC BY.
En la fotografía con lámpara de hendidura, se observan opacidades difusas grisáceas y depósitos maculares desde el centro de la córnea hasta toda la córnea. Muestra los hallazgos clínicos típicos de la distrofia corneal macular, facilitando la comprensión de las opacidades corneales que causan la pérdida de visión.
Pérdida de visión: Es la queja más frecuente. A menudo se nota entre los 10 y 30 años de edad 8)
Dolor ocular o irritación: Puede ocurrir con la erosión epitelial corneal recurrente
Fotofobia (sensibilidad a la luz): La función visual disminuye significativamente en ambientes brillantes a medida que progresan las opacidades corneales
Erosión epitelial corneal recurrente: Puede ocurrir repetidamente debido a una adhesión anormal del epitelio corneal
El patrón de progresión típico de los hallazgos clínicos es el siguiente.
Clínicamente, se observan depósitos finos difusos en el estroma corneal, causando una opacidad en vidrio esmerilado. A medida que progresa, la opacidad afecta todo el grosor del estroma y se extiende desde el centro hacia la periferia. Posteriormente, además de la opacidad tenue, aparecen numerosas opacidades pequeñas, irregulares y de color grisáceo en el estroma superficial a profundo. 9)
Hallazgos iniciales
Opacidades puntiformes: Aparecen pequeñas opacidades puntiformes grisáceas en el estroma superficial de la córnea central
Opacidad en vidrio esmerilado: Se observa una opacidad tenue difusa en el estroma corneal
Bordes indistintos: Los márgenes de las opacidades son poco claros y el límite con el estroma normal es indistinto
Hallazgos en etapa avanzada
Afectación de todo el espesor: La opacidad se extiende a través de todo el estroma
Extensión periférica: La opacidad se extiende desde el centro hacia la periferia
Adelgazamiento corneal: Disminuye el grosor corneal central
Depósitos endoteliales y en la membrana de Descemet: Se acumula material anormal en las estructuras profundas
Con el microscopio de lámpara de hendidura, toda la córnea aparece difusamente opaca con depósitos irregulares de color blanco grisáceo. Al realizar un corte óptico con la luz de hendidura, se observa una distribución característica: los depósitos se localizan en la capa superficial en el centro y en la capa profunda en la periferia. Las lesiones parcheadas suelen aparecer en un patrón concéntrico8).
La opacidad puede extenderse al limbo, lo que es un punto de diferenciación importante con otras distrofias corneales. En la distrofia corneal granular y la distrofia corneal reticular, el limbo suele permanecer claro, mientras que en la MCD, toda la córnea, incluido el limbo, es frecuentemente opaca2,8). El astigmatismo irregular se asocia con depósitos estromales anteriores, y también puede haber disminución de la sensibilidad corneal. Dado que el material anormal se deposita en el endotelio, los casos avanzados pueden desarrollar edema estromal debido a la disfunción endotelial8).
El curso natural varía entre individuos, pero a menudo sigue estas etapas:
Primera infancia (etapa asintomática): La mutación genética está presente desde el nacimiento, pero los hallazgos en la lámpara de hendidura son mínimos y el paciente es asintomático.
Edad escolar a adolescencia (etapa de opacidad temprana): Aparecen opacidades difusas tenues en el estroma corneal superficial, y eventualmente se hacen visibles depósitos parcheados.
10 a 30 años (etapa de disminución de la visión): La opacidad progresa y los pacientes notan una disminución de la visión.
30 a 40 años (etapa avanzada): La opacidad se extiende a todo el espesor del estroma y al limbo, con hipoestesia corneal, adelgazamiento y astigmatismo irregular evidentes.
Edad media a avanzada (etapa de indicación de trasplante de córnea): El deterioro visual alcanza un nivel que interfiere con la vida diaria y se considera el trasplante.
La MCD es una enfermedad progresiva con deterioro continuo de la función visual a lo largo de la vida, lo que difiere significativamente en el pronóstico de la distrofia corneal granular tipo I y algunos subtipos de distrofia corneal reticular, donde el deterioro visual permanece leve4,8).
El gen causante es CHST6 (carbohydrate sulfotransferase 6)3). Se localiza en 16q22 y codifica una enzima que transfiere grupos sulfato a la N-acetilglucosamina en los proteoglicanos corneales. Se han reportado muchos tipos de mutaciones en este gen en varios grupos étnicos, incluyendo mutaciones sin sentido, mutaciones sin sentido, mutaciones de cambio de marco y deleciones en la región 5’ corriente arriba3,7).
Debido a que es un trastorno autosómico recesivo, ambos padres de un individuo afectado suelen ser portadores. La incidencia tiende a ser mayor en regiones o poblaciones con matrimonios consanguíneos frecuentes. Los heterocigotos compuestos resultantes de matrimonios entre diferentes familias también pueden desarrollar la enfermedad7).
En 2000, Akama et al. identificaron CHST6 como el gen causante de esta enfermedad y demostraron que tanto el inmunofenotipo I como el II son causados por mutaciones en el mismo locus génico3). Este descubrimiento fue un hallazgo importante que sugiere que las diferencias en el inmunofenotipo están determinadas por diferentes patrones de mutación en un solo gen, y se convirtió en la base para los sistemas de diagnóstico genético posteriores.
Se han reportado más de 200 tipos de mutaciones, siendo las mutaciones sin sentido las más comunes. Sultana et al. identificaron muchas mutaciones novedosas en un grupo de pacientes del sur de la India, mostrando que la alta frecuencia en esa región se debe a una acumulación de mutaciones específicas de la región7). Se han reportado agrupaciones regionales similares en Arabia Saudita e Islandia, y se cree que los antecedentes históricos de la población (efectos fundadores y costumbres de matrimonios consanguíneos) contribuyen a la incidencia5,6,7).
La incidencia varía según la región, y la enfermedad es relativamente rara9). En comparación con otras distrofias corneales como la distrofia corneal granular, hay menos casos y tiende a reportarse en familias con heterocigosidad compuesta o antecedentes de matrimonios consanguíneos.
Antecedentes familiares: Debido a que es autosómico recesivo, ambos padres deben ser portadores.
Matrimonio consanguíneo: Aumenta la incidencia.
Región geográfica: Mayor prevalencia en el sur de la India, Arabia Saudita, Islandia y Escandinavia5,7).
Q¿Es necesaria la prueba genética?
A
La prueba genética del gen CHST6 es útil para el diagnóstico definitivo. Las pruebas genéticas pueden realizarse en instituciones médicas acreditadas. Sin embargo, en muchos centros, el diagnóstico clínico mediante microscopía de lámpara de hendidura es el método principal. Es útil para evaluar el riesgo de enfermedad en futuros hijos y para confirmar el diagnóstico en casos con hallazgos clínicos atípicos.
El desencadenante para sospechar MCD es “opacidad corneal bilateral, progresiva y difusa en adolescentes y adultos jóvenes”. Primero, se obtiene una historia detallada de los síntomas subjetivos (disminución de la visión, fotofobia, sensación de irritación), antecedentes familiares y matrimonio consanguíneo. Luego, el enfoque estándar incluye la evaluación de los hallazgos corneales con lámpara de hendidura, evaluación de la función endotelial y, si es necesario, pruebas genéticas.
Esta es la prueba básica para el diagnóstico. Si se observa opacidad corneal bilateral sin congestión ni edema corneal, se debe sospechar distrofia corneal. En la MCD, los siguientes hallazgos son característicos:
Opacidad difusa en vidrio esmerilado: Toda la córnea aparece turbia de forma difusa
Depósitos punteados: Numerosas opacidades irregulares de color gris blanquecino
Distribución concéntrica: Con el haz de hendidura, las opacidades centrales son superficiales, las periféricas son profundas
Afectación del limbo: Las opacidades pueden extenderse al limbo
Anomalías endoteliales: En casos avanzados, pueden observarse depósitos en forma de gota
Mientras que muchas distrofias corneales aparecen como lesiones discontinuas (con áreas claras entre los depósitos) a nivel de la lámpara de hendidura, la MCD presenta excepcionalmente un patrón de opacidad difusa. Junto con la distrofia corneal reticular tipo I y la distrofia corneal gelatinosa en gotas, se caracteriza como un ejemplo representativo de “depósitos corneales observados difusamente”.
Microscopía confocal in vivo: Muestra material altamente reflectante con bordes indistintos y pérdida de imágenes normales de queratocitos8)
Topografía corneal: Demuestra aumento de densidad en el ápice corneal y adelgazamiento corneal central
Microscopía ultrasónica biomicroscópica (UBM): Útil para evaluar opacidades profundas y estructuras corneales posteriores
Microscopio especular: Evaluación de la densidad y morfología de las células endoteliales. El grado de patología endotelial influye directamente en la selección de la técnica quirúrgica
Pentacam corneal (cámara Scheimpflug): Proporciona un mapa de densidad de todo el espesor corneal, útil para la evaluación tridimensional de las opacidades
Histológicamente, muestra positividad con tinción de azul alcián y hierro coloidal, y se observa acumulación difusa de glucosaminoglicanos hiposulfatados dentro y alrededor de los queratocitos en el estroma corneal2,8). Pueden observarse roturas en la membrana de Bowman, y en casos avanzados, también se encuentra material anormal dentro de las células endoteliales. Pueden presentarse hallazgos similares a guttata en la membrana de Descemet.
Cristales en forma de aguja, asociado con hiperlipidemia
Distrofia corneal estromal congénita
Autosómica dominante
Presente al nacer, engrosamiento estromal
Mucopolisacaridosis sistémica (tipo corneal)
Recesiva/ligada al X
Asociado con síntomas sistémicos
Otras condiciones a considerar incluyen la distrofia corneal amorfa posterior (PACD) y la distrofia corneal pre-Descemet (PDCD). Las mucopolisacaridosis sistémicas (como los síndromes de Hurler, Scheie y Morquio) también pueden presentar opacidad corneal, por lo que es necesaria una evaluación que incluya hallazgos sistémicos 8).
Q¿Cómo se diagnostica la distrofia corneal macular?
A
El diagnóstico clínico se realiza mediante microscopía con lámpara de hendidura que muestra opacidad corneal difusa en vidrio esmerilado y depósitos maculares, naturaleza bilateral y progresiva, antecedentes familiares y edad de inicio (10-30 años). La prueba genética de CHST6 es útil para el diagnóstico definitivo.
Los objetivos del tratamiento para la MCD son: (1) mantener y recuperar la función visual, (2) aliviar el dolor y los síntomas de irritación mediante la estabilización de la superficie ocular, y (3) prevenir complicaciones (erosión epitelial, infección). Dado que no existe un tratamiento etiológico fundamental, se realizan intervenciones escalonadas según la etapa de la enfermedad y los síntomas.
No se ha establecido una terapia farmacológica para suprimir la progresión de esta enfermedad 8). Se realizan las siguientes medidas para el alivio de los síntomas.
Lágrimas artificiales: Protección de la superficie ocular y prevención de la sequedad
En casos con pérdida visual progresiva, el trasplante de córnea es la única opción curativa. La técnica quirúrgica se elige según la presencia o ausencia de afectación endotelial.
Queratoplastia lamelar anterior profunda (DALK): Es la primera opción en casos sin afectación endotelial 8,9). Al preservar el endotelio corneal propio del paciente, el riesgo de rechazo del injerto es menor que en la queratoplastia penetrante. El informe de evaluación de la Academia Americana de Oftalmología (AAO) también indica que, para las distrofias estromales, la DALK logra una recuperación visual equivalente a la QP con menor pérdida endotelial 8). La tasa de recurrencia después de DALK se reporta como baja, y es poco probable que ocurra recurrencia porque el estroma corneal del receptor se reemplaza.
Queratoplastia penetrante (QP): Está indicada en casos avanzados con depósitos anormales que afectan el endotelio y la membrana de Descemet, y en casos con adelgazamiento corneal central severo 7). La edad promedio para la primera QP por MCD se reporta entre los 30 y 40 años 7), y la supervivencia del injerto es buena.
Queratectomía fototerapéutica (PTK): Se realiza sintomáticamente para la erosión epitelial corneal recurrente y las opacidades cicatriciales superficiales. Sin embargo, se debe tener precaución con la hipermetropía inducida y la opacidad estromal.
En la MCD, los depósitos anormales también pueden afectar las células endoteliales, por lo que se tiende a elegir la QP sobre la DALK cuando la enfermedad se extiende profundamente7). El trasplante de córnea de espesor total está indicado cuando hay anomalías endoteliales. La evaluación endotelial preoperatoria detallada (microscopía especular y confocal) es clave para determinar la técnica quirúrgica.
La tasa de recurrencia después de DALK es baja, reportada en aproximadamente un 5%. La supervivencia del injerto después de PKP es buena, con muchos informes que logran supervivencia a largo plazo; sin embargo, también se han reportado casos de redepósito de material anormal en el injerto varios años o más de una década después de la cirugía 8). En la serie de Arabia Saudita de Al-Swailem et al., la supervivencia del injerto después de PKP para MCD fue buena, pero se observó recurrencia en algunos casos durante el seguimiento a largo plazo 8). Además, la revisión de la AAO por Reinhart et al. mostró que DALK logra una recuperación visual igual o mejor que PKP y una baja pérdida de células endoteliales para las distrofias estromales 9). Un estudio multicéntrico de Unal et al. también reportó la eficacia de DALK para distrofias estromales, incluida MCD.
En general, la recurrencia postoperatoria de los injertos corneales ocurre porque la patología molecular de la enfermedad original permanece en el huésped. En DALK, se conservan el endotelio del huésped y la capa pre-Descemet, por lo que en casos con afectación endotelial, la enfermedad puede progresar. Es necesario un seguimiento regular a largo plazo (agudeza visual, examen con lámpara de hendidura, medición de la densidad de células endoteliales) después de la cirugía.
Q¿Cuál debo elegir, DALK o PKP?
A
Si el endotelio y la membrana de Descemet no están afectados, la queratoplastia lamelar anterior profunda (DALK) es la primera opción. DALK preserva el endotelio corneal propio del paciente, por lo que el riesgo de rechazo es bajo y la tasa de recurrencia postoperatoria se reporta en aproximadamente un 5%. Por otro lado, en casos donde los depósitos anormales también afectan al endotelio, está indicada la queratoplastia penetrante (PKP). El procedimiento quirúrgico se determina según la evaluación endotelial preoperatoria.
El gen CHST6 codifica la carbohidrato sulfotransferasa 6 3). Esta enzima es responsable de transferir grupos sulfato a la N-acetilglucosamina en las moléculas de queratán y es esencial para la síntesis normal del sulfato de queratán (KS) en los proteoglicanos corneales.
Cuando la actividad enzimática se pierde debido a una mutación genética, se sintetiza sulfato de queratán hiposulfatado. Este sulfato de queratán anormal tiene baja solubilidad y se acumula de manera anormal dentro y fuera de los queratocitos en el estroma corneal2,3).
Las anomalías cuantitativas y cualitativas del sulfato de queratán conducen a la siguiente cadena de cambios patológicos.
Producción anormal de proteoglicanos pequeños ricos en leucina (SLRP): Los SLRP específicos de la córnea, como lumican, keratocan y mimecan, no se sintetizan normalmente.
Anomalía en la disposición de las fibras de colágeno: Estos SLRP controlan estrictamente el diámetro y el espaciado de las fibras de colágeno corneal, asegurando la transparencia. La disminución de la función de los SLRP provoca diámetros desiguales de las fibras de colágeno y alteración del espaciado2).
Acumulación anormal en la matriz extracelular: El queratán no sulfatado se deposita en la matriz extracelular.
Aumento de la dispersión de la luz y pérdida de transparencia: Los cambios combinados anteriores aumentan la dispersión de la luz visible, causando opacidad difusa en toda la córnea.
La acumulación de glucosaminoglicanos se observa dentro y fuera de los queratocitos del estroma y, a medida que la enfermedad progresa, se extiende a la membrana de Bowman, la membrana de Descemet y las células endoteliales. En el tipo I, también se ha confirmado una actividad enzimática reducida en el cartílago auricular, lo que sugiere que podría ser una manifestación parcial de una anomalía sistémica del metabolismo del sulfato de queratán2). Sin embargo, los síntomas sistémicos son raros clínicamente y se trata como una enfermedad localizada con afectación corneal principal.
Entre los proteoglicanos corneales, el lumican controla el diámetro de las fibras de colágeno a aproximadamente 25 nm, mientras que el keratocan y el mimecan mantienen un espaciado uniforme. Cuando las cadenas laterales sulfatadas de estos SLRP son cortas e incompletas, las fibras de colágeno se vuelven variables en grosor y el espaciado se vuelve desigual. Como resultado, aumenta la dispersión de la luz dentro del estroma corneal, observándose clínicamente como opacidad en vidrio esmerilado.
Clásicamente, la teoría reticular de Maurer explica la transparencia corneal mediante la cancelación de la interferencia de la luz debido a la disposición reticular ordenada de las fibras de colágeno. En la MCD, esta estructura reticular se altera por anomalías de los SLRP, lo que lleva a la pérdida de transparencia2,4).
Investigaciones básicas recientes han informado que la disfunción de la autofagia debida a mutaciones en CHST6 puede inducir piroptosis (muerte celular inflamatoria) de los queratocitos, contribuyendo a la progresión de la enfermedad8). Anomalías similares de la autofagia se han informado en otras distrofias corneales (como la granular tipo II), atrayendo la atención como una patología común en las distrofias corneales.
7. Investigación más reciente y perspectivas futuras
La terapia génica dirigida se ha propuesto como una estrategia de tratamiento permanente 8). La investigación básica sobre edición genética con CRISPR/Cas9 está avanzando en la distrofia epitelial corneal de Meesmann, y podría convertirse en una opción terapéutica futura para la MCD. Sin embargo, quedan muchos desafíos para la aplicación clínica, como la edición no deseada de alelos normales (efectos fuera del objetivo), el establecimiento de métodos eficientes de transferencia génica a los queratocitos y la verificación de la seguridad a largo plazo.
Se está investigando a nivel básico un enfoque para eliminar enzimáticamente el sulfato de queratán hiposulfatado acumulado en la córnea. Esto aplica el concepto de terapia de reemplazo enzimático, ya utilizado en mucopolisacaridosis sistémicas, a la córnea local, pero actualmente no hay informes de aplicación clínica.
La queratoplastia endotelial automatizada con pelado de la membrana de Descemet (DSAEK) y la queratoplastia endotelial de la membrana de Descemet (DMEK) son técnicas quirúrgicas desarrolladas principalmente para la distrofia endotelial corneal de Fuchs y la queratopatía bullosa, pero su aplicación en casos de MCD con afectación endotelial temprana es un tema de investigación futura. Actualmente, la queratoplastia penetrante (PKP) es la opción realista para esta enfermedad, que afecta tanto al estroma como al endotelio. En el futuro, se ha propuesto el concepto de “queratoplastia secuencial” que combina la queratoplastia lamelar anterior profunda (DALK), que reemplaza solo el estroma, y la DMEK, que reemplaza solo el endotelio, en etapas, pero todas están en fase de investigación.
Se ha informado que los niveles séricos de sulfato de queratán son más bajos de lo normal en pacientes con MCD de inmunofenotipo tipo II, y se está discutiendo su utilidad como marcador metabólico sistémico 2). En el futuro, se espera su aplicación en el cribado mediante análisis de sangre y en el diagnóstico temprano de portadores en familias con mutaciones en CHST6. Además, el análisis automatizado de imágenes de lámpara de hendidura mediante inteligencia artificial es un área de investigación que puede contribuir a la detección temprana de enfermedades raras como esta.
Dado que es un trastorno autosómico recesivo, aproximadamente el 25% de los hermanos del paciente pueden estar afectados. Las pruebas genéticas de CHST6 permiten el cribado familiar y el asesoramiento genético sobre el matrimonio y el embarazo. Se recomienda la derivación temprana a un especialista para familias con antecedentes de matrimonio consanguíneo o casos de opacidad corneal similar dentro de la familia.
Akama TO, Nishida K, Nakayama J, et al. Macular corneal dystrophy type I and type II are caused by distinct mutations in a new sulphotransferase gene. Nature Genetics. 2000;26(2):237-241. doi:10.1038/79987.
Aggarwal S, Peck T, Golen J, Karcioglu ZA. Macular corneal dystrophy: A review. Survey of Ophthalmology. 2018;63(5):609-617. doi:10.1016/j.survophthal.2018.03.004.
Jonasson F, Oshima E, Thonar EJ, et al. Macular corneal dystrophy in Iceland. A clinical, genealogic, and immunohistochemical study of 28 patients. Ophthalmology. 1996;103(7):1111-1117. doi:10.1016/s0161-6420(96)30559-9.
Jonasson F, Johannsson JH, Garner A, Rice NS. Macular corneal dystrophy in Iceland. Eye (London, England). 1989;3 ( Pt 4):446-54. doi:10.1038/eye.1989.66. PMID:2606219.
Sultana A, Sridhar MS, Jagannathan A, et al. Novel mutations of the carbohydrate sulfotransferase-6 (CHST6) gene causing macular corneal dystrophy in India. Mol Vis. 2003;9:730-734. PMID:14735064.
Al-Swailem SA, Al-Rajhi AA, Wagoner MD. Penetrating keratoplasty for macular corneal dystrophy. Ophthalmology. 2005;112(2):220-224. doi:10.1016/j.ophtha.2004.08.017.
Reinhart WJ, Musch DC, Jacobs DS, Lee WB, Kaufman SC, Shtein RM. Deep anterior lamellar keratoplasty as an alternative to penetrating keratoplasty a report by the american academy of ophthalmology. Ophthalmology. 2011;118(1):209-218. doi:10.1016/j.ophtha.2010.11.002. PMID:21199711.
Copia el texto del artículo y pégalo en el asistente de IA que prefieras.
Artículo copiado al portapapeles
Abre un asistente de IA abajo y pega el texto copiado en el chat.