A distrofia corneana reticular (lattice corneal dystrophy, LCD) é uma distrofia corneana hereditária na qual a amiloide se deposita no estroma corneano, causando opacidades lineares em forma de treliça. Descrita na década de 1890, é uma doença de longa data. Na segunda edição da classificação clínico-genética da IC3D (International Committee for Classification of Corneal Dystrophies), é integrada como LCD1 e suas variantes (tipos 3, 3A, 1/3A e 4 anteriores)4).
Posicionamento como distrofia relacionada ao TGFBI
LCD1, distrofia corneana granular, distrofia corneana de Reis-Bücklers e distrofia corneana de Thiel-Behnke formam um grupo de doenças como “distrofias relacionadas ao TGFBI”. O gene causador TGFBI (transforming growth factor beta-induced gene) está localizado no braço longo do cromossomo 5 (5q31) e segue herança autossômica dominante. A proteína TGFBI (TGFBIp, kerato-epithelin, βig-h3) é produzida pelo epitélio corneano e distribuída por toda a espessura da córnea. No estroma corneano, está envolvida na organização das fibrilas de colágeno. Mesmo com mutações no mesmo gene, diferenças no local da mutação e no aminoácido substituído resultam em grande diferenciação no material depositado (hialina ou amiloide) e no quadro clínico5).
A mutação representativa do LCD1 é a R124C, onde a arginina na posição 124 do gene TGFBI é substituída por cisteína. Na variante LCD IIIA, mutações como L527R foram relatadas.
A proteína anormal acumulada na córnea cora-se de vermelho com vermelho Congo e exibe birrefringência verde-maçã característica ao microscópio de polarização, confirmando-se como amiloide. Este achado é um indicador clássico de diagnóstico tecidual de amiloidose desde o século XIX6).
O tipo anteriormente chamado de “distrofia corneana em treliça tipo 2” é uma manifestação ocular da amiloidose sistêmica por gelsolina (GSN-AMYL, síndrome de Meretoja). Na classificação atual do IC3D, é classificado como “amiloidose familiar” e tratado separadamente do LCD clássico4,10). Descrita por Meretoja em 1969 na Finlândia, esta síndrome é uma doença hereditária que, além da opacidade corneana em treliça, inclui neuropatia craniana progressiva, flacidez cutânea e sintomas sistêmicos10,11). Como a diferenciação entre ambas é importante na prática clínica, este artigo também as descreve em conjunto.
Opacidades filiformes de contorno duplo na área pupilar, erosões epiteliais recorrentes
LCD IIIA (tipo variante)
TGFBI (5q31)
L527R, etc.
Após os 40 anos
Linhas reticulares grossas em corda no estroma profundo, sem alterações epiteliais
Tipo GSN (Meretoja)
GSN (9q34)
D187N, p.Glu580Lys2)
30-40 anos
Linhas reticulares radiais na periferia, amiloidose sistêmica
No Japão, a distrofia relacionada ao TGFBI mais frequente é esmagadoramente a granular tipo II (tipo Avellino, R124H), e a LCD1 é menos comum em comparação. No entanto, como ambas se diferenciam por apenas alguns nucleotídeos no mesmo gene TGFBI, em casos com sobreposição de quadros clínicos, é desejável a confirmação por teste genético. A prevalência exata de LCD no Japão não foi relatada, mas está entre as relativamente raras dentro do conjunto das distrofias corneanas.
QQual a diferença entre LCD1 e síndrome de Meretoja?
A
A LCD1 é um depósito amiloide limitado à córnea devido a mutações no gene TGFBI, iniciando-se na área pupilar entre 10 e 20 anos de idade e frequentemente acompanhada de erosões epiteliais recorrentes. Por outro lado, a síndrome de Meretoja (antiga LCD2, tipo GSN) é uma manifestação ocular de amiloidose sistêmica causada por mutações no gene GSN (gelsolina), iniciando-se na periferia da córnea entre 30 e 40 anos, com transparência central mantida por longo tempo. A síndrome de Meretoja apresenta sintomas sistêmicos como flacidez cutânea, fácies em máscara, neuropatia periférica e arritmias cardíacas2,10). Na 2ª edição do IC3D, a síndrome de Meretoja é classificada independentemente da distrofia corneana reticular4).
Liu Z, et al. An R124C mutation in TGFBI caused lattice corneal dystrophy type I with a variable phenotype in three Chinese families. Mol Vis. 2008. Figure 2. PMCID: PMC2443752. License: CC BY.
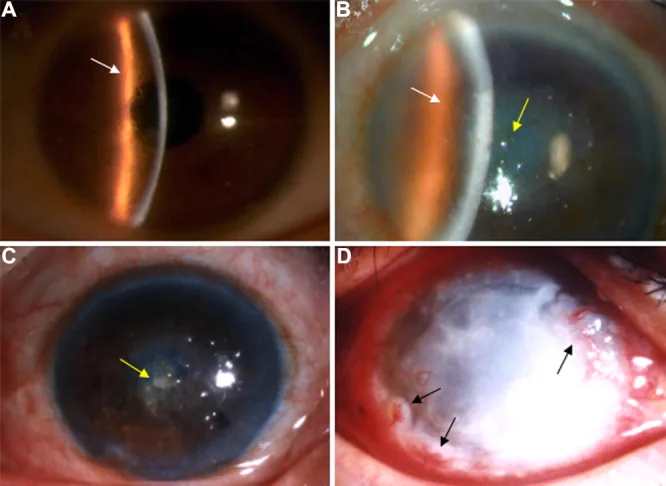
Na fotografia com lâmpada de fenda, observam-se linhas em treliça ramificadas no estroma corneano e opacidade predominante central. Esta imagem mostra os achados clínicos típicos da distrofia corneana em treliça.
No LCD1, muitos pacientes são assintomáticos na infância, apresentando apenas opacidades finas detectáveis pelo método de retroiluminação da lâmpada de fenda. A partir da segunda ou terceira década de vida, ocorrem erosões epiteliais recorrentes (RCE), com dor ocular intensa ao acordar, fotofobia, lacrimejamento e sensação de corpo estranho. Por volta dos 30 anos, opacidades brancas tornam-se evidentes no estroma anterior central da córnea, e a acuidade visual diminui progressivamente após os 40 anos.
No LCD IIIA (tipo variante), geralmente não ocorre dano epitelial, e a queixa principal é a diminuição lenta da visão após os 40 anos.
No antigo LCD2 (síndrome de Meretoja), os sintomas oculares aparecem entre os 30 e 40 anos, mas o comprometimento visual significativo geralmente é adiado até os 60 anos11). Frequentemente, sintomas sistêmicos como flacidez da pele palpebral, fácies em máscara, neuropatia craniana progressiva e arritmia cardíaca precedem ou acompanham os sintomas oculares2,10).
Local de início: Aparece como opacidades puntiformes e lineares finas na camada de Bowman e estroma anterior na área pupilar de ambos os olhos.
Linhas em treliça: Opacidades filamentosas ou lineares com contorno duplo entrelaçam-se, formando opacidades em rede ou estreladas.
Fase avançada: Opacidade branco-leitosa oval ou circular na córnea central.
Retroiluminação: Linhas finas em treliça, pouco visíveis na iluminação direta, tornam-se claramente evidentes.
Coloração com fluoresceína: A adesão epitelial reduzida torna a superfície áspera.
Erosão epitelial recorrente: Ocorre com alta frequência porque os depósitos atingem as células basais do epitélio e a membrana de Bowman.
LCD IIIA (tipo variante)
Linhas em grade: Linhas em grade espessas e longas no estroma médio a profundo, às vezes com ramificação dendrítica. Observáveis mesmo com iluminação direta.
Fenótipo: Existem três padrões: ① apenas linhas em grade, ② apenas depósitos granulares pequenos, ③ mistura de ambos. Podem ocorrer fenótipos diferentes entre os olhos direito e esquerdo no mesmo indivíduo, ou casos unilaterais.
Epitélio: Normalmente, não ocorre dano epitelial.
Homozigoto: Em homozigotos L527R, as linhas em grade são mais espessas e os depósitos granulares centrais são maiores, mas a diferença não é tão marcante quanto entre hetero e homozigotos para R124H (tipo granular II).
Tipo GSN (Meretoja)
Linhas em grade: Poucos depósitos em grade, menos delicados, aparecendo radialmente a partir da periferia.
Transparência central: A transparência central é mantida por um longo período após o início.
Erosão epitelial: Rara.
Achados sistêmicos: Apresentam alterações faciais como fácies em máscara, lábios proeminentes com distúrbios motores, orelhas caídas, e blefarocalásia 2).
No LCD1, alguns casos apresentam opacidade circular central particularmente intensa; há relato de um heterozigoto R124C de 56 anos que necessitou de transplante de córnea devido a opacidade circular central.
QUma criança pode ser diagnosticada com LCD1?
A
Na infância, o LCD1 é frequentemente assintomático e difícil de detectar apenas com iluminação direta. Com observação detalhada usando iluminação de transiluminação ou retroiluminação do biomicroscópio, é possível confirmar opacidades puntiformes a lineares finas no estroma superficial central. Em crianças com erosões epiteliais recorrentes, recomenda-se considerar LCD1, incluindo histórico familiar e exame da córnea dos pais. O teste genético TGFBI é útil para o diagnóstico definitivo.
Os genes causadores e mutações representativas da distrofia corneana em grade estão organizados abaixo.
Relacionado ao TGFBI (LCD1, LCD IIIA, LCD IV)
Locus gênico: 5q31 (gene TGFBI).
Padrão de herança: Autossômico dominante.
Mutação representativa da LCD1: R124C (Arg124Cys) é a mais frequente5).
Mutação representativa da LCD IIIA: L527R (Leu527Arg) foi relatada, entre outras. Também existem casos homozigotos.
Mutação de novo: Foi relatado um caso com mutação de novo L509P no TGFBI que apresentou fenótipo de LCD IIIA1). Os pais não apresentavam a mutação, mas um dos filhos a herdou1).
Papel da TGFBIp: Produzida pelo epitélio corneano, distribui-se por toda a espessura da córnea e, no estroma, participa da organização das fibras de colágeno5).
Relacionado ao GSN (Síndrome de Meretoja, antiga LCD2)
Locus gênico: 9q34 (gene GSN, gelsolina).
Padrão de herança: Autossômico dominante.
Mutação clássica: D187N (tipo finlandês) é a mais comum; p.Asp187Tyr também foi relatada10,11).
Nova mutação: p.Glu580Lys, relatada em uma família eslovena, localiza-se na fronteira dos domínios G4-G5 e causa repulsão eletrostática devido à substituição de carga negativa por positiva2).
Quadro clínico: Além da opacidade em treliça da córnea, apresenta amiloidose sistêmica com flacidez cutânea, arritmia cardíaca, insuficiência renal e neuropatia óptica2).
Por ser uma doença hereditária, o histórico familiar é o fator de risco mais importante. No entanto, como mutações de novo podem ocorrer no TGFBI, a ausência de histórico familiar não exclui a doença1). O padrão de herança é autossômico dominante; se um dos pais for portador da mutação, há 50% de chance de transmissão para os filhos. Não há diferença entre sexos, e a diferença racial não é clara na LCD1, mas a síndrome de Meretoja é conhecida por se concentrar em famílias numerosas na Finlândia11).
A contribuição de fatores ambientais não é clara; o início e a progressão da doença são determinados principalmente pelo genótipo. No entanto, a frequência de erosões epiteliais recorrentes pode aumentar em ambientes secos, uso de lentes de contato ou trauma. Cirurgias refrativas (LASIK, SMILE, etc.) podem causar rápida deterioração das distrofias associadas ao TGFBI, sendo necessária atenção em casos com histórico familiar durante a triagem pré-operatória5).
Para diferenciar LCD1, suas variantes e o tipo GSN, é necessário integrar os achados da lâmpada de fenda, histológicos e genéticos.
Exames clínicos
Microscopia com lâmpada de fenda: Na iluminação direta, as linhas reticulares iniciais são facilmente negligenciadas. Com a técnica de transiluminação, detectam-se opacidades finas contra o fundo pupilar; com a iluminação de retroiluminação, detectam-se linhas reticulares finas e semitransparentes.
Coloração com fluoresceína: Na LCD1, devido à redução da adesão epitelial, a coloração apresenta aspecto rugoso. Também é útil para avaliar a extensão da erosão epitelial.
Microscopia confocal da córnea: Permite observar os depósitos no estroma em nível celular.
Diagnóstico definitivo
Teste genético: A detecção de mutações nos genes TGFBI e GSN confirma o tipo da doença. Como mutações diferentes, mesmo com o mesmo fenótipo, alteram a velocidade de recorrência e progressão, isso impacta diretamente o plano de tratamento.
Exame anatomopatológico: A coloração com vermelho Congo mostra coloração vermelha e, sob microscopia de polarização, exibe birrefringência verde-maçã, confirmando a presença de amiloide6).
Imuno-histoquímica: A diferenciação entre os tipos é possível com anticorpos anti-TGFBIp e anti-gelsolina.
História familiar: Como a herança é autossômica dominante, a confirmação dos achados corneanos em pais e irmãos apoia o diagnóstico.
Distrofia corneana granular tipo II (tipo Avellino, TGFBI R124H): É a distrofia relacionada ao TGFBI mais frequente no Japão, apresentando um padrão misto de depósitos granulares e linhas reticulares. Para diferenciar da LCD1, o teste genético é definitivo.
Amiloidose corneana secundária: não hereditária, a amiloide deposita-se secundariamente a estímulos crônicos da superfície ocular, como triquíase e ceratocone. A ausência de história familiar e a presença de doença de base são pontos de diferenciação.
Distrofia corneana macular: herança autossômica recessiva por mutação no gene CHST6, com opacidade difusa em vidro fosco e anormalidades endoteliais.
Distrofia corneana gelatinosa em gotas: herança autossômica recessiva por mutação no gene TACSTD2, apresentando elevações gelatinosas esbranquiçadas. Relativamente comum no Japão.
QPor que o teste genético é importante?
A
Na distrofia corneana em treliça, mesmo com fenótipos semelhantes, genes causadores e locais de mutação diferentes alteram significativamente a velocidade de progressão, frequência de recorrência, opções de tratamento e presença de complicações sistêmicas. A LCD1 por mutação no TGFBI e a síndrome de Meretoja por mutação no GSN diferem fundamentalmente na abordagem terapêutica e na necessidade de investigação sistêmica2,10). Além disso, há relatos de mutações de novo que impossibilitam a determinação do tipo apenas pela história familiar1), tornando o teste genético essencial para o diagnóstico definitivo e a classificação do subtipo.
Na infância e juventude, quando assintomática ou com apenas opacidades finas, a conduta é observação. Avaliar a progressão com exame de lâmpada de fenda a cada 6 meses a 1 ano.
Para a erosão epitelial recorrente, sintoma central da LCD1, o primeiro passo é o tratamento conservador abaixo.
Tratamento da crise: uso contínuo de lentes de contato gelatinosas terapêuticas para proteger o epitélio corneano. Associar colírios antibióticos para prevenir infecção secundária. Aplicar pomada oftálmica para lubrificação e proteção epitelial.
Prevenção de recorrência: a aplicação de pomada oftálmica ao deitar tem efeito na redução de crises de RCE. Em ambientes secos, usar lágrimas artificiais ou lubrificantes também durante o dia.
Na LCD1, onde o depósito amiloide é predominantemente na camada superficial da córnea, em casos de opacidade central intensa ou erosão epitelial recorrente frequente, a ceratectomia fototerapêutica (PTK) com laser excimer é a primeira escolha7,8). Geralmente não há recorrência precoce, mas a recidiva ao longo do tempo é inevitável; o tratamento com PTK pode ser realizado até cerca de duas vezes no mesmo olho.
Em heterozigotos, a recorrência é lenta e poucos casos necessitam de retratamento. Em homozigotos, há tendência a recorrência mais precoce em comparação com heterozigotos. A taxa de recorrência após PTK aumenta com o tempo, semelhante a outras distrofias relacionadas ao TGFBI, e na observação de longo prazo, a maioria dos casos apresenta algum sinal de recorrência8).
Como um caso demonstrando a eficácia da PTK, em um caso de LCD IIIA devido à mutação de novo TGFBI L509P, foi realizada PTK guiada por FD-OCT com 60 µm, e a melhor acuidade visual corrigida (BCVA) melhorou de 20/400 para 20/501). Aos 45 meses de pós-operatório, não foi observada diminuição da acuidade visual ou recorrência significativa1).
De acordo com o Preferred Practice Pattern da AAO para edema e opacidade corneana, a PTK para distrofias corneanas granulares e em grade é uma “opção razoável” e pode retardar a transição para DALK ou transplante de córnea total, mas há risco de haze pós-operatório. Quando repetida, o uso combinado de mitomicina C é considerado como meio de suprimir cicatrizes recorrentes e depósitos estromais. É alertado que o risco de ectasia corneana aumenta quando a ablação ultrapassa o terço anterior do estroma ou quando o leito residual é inferior a 250 µm7).
Em casos de recorrência repetida ou quando a opacidade atinge camadas mais profundas do estroma, opta-se pelo transplante de córnea. Na LCD1, geralmente não há indicação de transplante de córnea até após os 40 anos. Na LCD, as células endoteliais da córnea são normalmente normais, portanto, a técnica cirúrgica é escolhida de acordo com a profundidade da opacidade.
Alta recuperação visual, mas com risco de rejeição e recorrência
Nos últimos anos, devido à redução do risco de rejeição e aos resultados visuais comparáveis aos do transplante de córnea de espessura total, a DALK tem sido amplamente utilizada como nova primeira escolha.
A recorrência da LCD após transplante de córnea é um fenômeno inevitável, com taxas de recorrência após transplante de espessura total relatadas como 17,8% em 5 anos, 26% em 8 anos e 56% em 15 anos9). Como a opacidade recorrente geralmente se limita à camada superficial, pode ser removida por PTK, prolongando o intervalo até o retransplante. Para LCD IIIA (tipo variante), muitas vezes não é necessário tratamento, a menos que haja impacto significativo na visão.
QQuão eficaz é a PTK?
A
A PTK pode remover efetivamente os depósitos de amiloide superficiais, melhorando a visão e reduzindo erosões epiteliais recorrentes. Em um caso de LCD IIIA, a melhor acuidade visual corrigida melhorou de 20/400 para 20/50 após PTK de 60 µm, sem recorrência por 45 meses1). Em heterozigotos, a recorrência é lenta, mas em homozigotos ocorre recorrência precoce. Lesões profundas não podem ser removidas por PTK, portanto, opacidades profundas requerem DALK ou transplante de córnea de espessura total7).
QA LCD recorre após o transplante de córnea?
A
A recorrência da LCD após transplante de córnea é inevitável. As taxas de recorrência após transplante de espessura total são relatadas como 17,8% em 5 anos, 26% em 8 anos e 56% em 15 anos9). No entanto, como a opacidade recorrente geralmente se limita à camada superficial do enxerto, pode ser removida por PTK, prolongando a vida útil do enxerto. A ceratoplastia lamelar anterior profunda (DALK) apresenta menor risco de rejeição endotelial em comparação com o transplante de espessura total e tem ganhado destaque como nova primeira escolha7).
6. Fisiopatologia e mecanismos detalhados da doença
O centro da patologia da LCD1 é o acúmulo anormal de TGFBIp (kerato-epitelina, βig-h3). O TGFBIp é normalmente produzido pelo epitélio corneano e distribuído por toda a espessura da córnea, sendo uma proteína estrutural envolvida na montagem das fibrilas de colágeno e na adesão celular no estroma5). A proteína anormal produzida pela mutação R124C sofre misfolding e autoagregação, depositando-se como fibrilas amiloides insolúveis na camada de Bowman e no estroma superficial. Em estágios avançados, os depósitos se espalham para o estroma profundo.
A deposição amiloide causa alterações nas estruturas de adesão epitelial da córnea anterior, levando à degeneração das células basais do epitélio e à degeneração da camada epitelial com defeitos na membrana de Bowman. Essa ruptura estrutural constitui a base patogênica das erosões epiteliais recorrentes da córnea.
Diferenciação fenotípica de acordo com o genótipo do TGFBI
No gene TGFBI, a diferença no local da mutação e no aminoácido substituído determina o quadro clínico. R124C causa LCD1, R124H causa distrofia corneana granular tipo II (tipo Avellino) e R124L causa distrofia corneana de Reis-Bücklers5). O mecanismo molecular pelo qual uma diferença de apenas um aminoácido determina o material depositado (amiloide vs. hialino vs. ambos) e o local de deposição ainda não foi completamente elucidado, mas acredita-se que o domínio do βig-h3 ao qual pertence o local da mutação e seu efeito na estabilidade do dobramento sejam fundamentais.
Na LCD IIIA, mutações de predomínio profundo, como L527R, produzem linhas lattice grossas em forma de corda, resultando em uma forma de início tardio sem envolvimento epitelial. A localização estratificada dos depósitos pode ser explicada pelo gradiente de secreção e difusão do βig-h3 das células produtoras (células basais do epitélio) para o estroma, juntamente com diferenças na estabilidade do dobramento da proteína mutante. Acredita-se que a R124C favoreça a via de formação de fibrilas amiloides a partir de intermediários de dobramento, acumulando amiloide ao redor da camada de Bowman5). Por outro lado, a mutação L527R forma uma proteína misfoldada relativamente estável, depositando-se lentamente nas camadas mais profundas do estroma.
Amiloide corneano posterior e risco cirúrgico intraocular
Tradicionalmente, acreditava-se que a deposição amiloide na LCD1 estava limitada à córnea anterior (camada de Bowman ao estroma superficial). No entanto, estudos patológicos recentes demonstraram a presença de depósitos amiloides também na córnea posterior, próximo à membrana de Descemet3). A deposição amiloide na córnea posterior pode afetar a adesão da membrana de Descemet e contribuir para o descolamento da membrana de Descemet durante a cirurgia de catarata3). Sugere-se que um mecanismo semelhante ao que prejudica a adesão epitelial na córnea anterior também atue na região posterior3).
A gelsolina, molécula causadora da antiga LCD2 (síndrome de Meretoja), está presente tanto no citoplasma quanto no espaço extracelular e é uma proteína envolvida na motilidade celular, divisão celular e apoptose através da ligação à actina. A mutação clássica D187N, conhecida como tipo finlandês, apresenta um fenótipo caracterizado por depósitos corneanos em treliça e neuropatia craniana11). A nova mutação p.Glu580Lys, relatada em uma família eslovena, está localizada na fronteira dos domínios G4-G5 e, pela substituição do glutamato de carga negativa pela lisina de carga positiva, gera repulsão eletrostática, reduzindo a conectividade e estabilidade entre os domínios2). A gelsolina mutante sofre clivagem anormal no plasma por furina e MT1-MMP, liberando fragmentos precursores de amiloide de 8 kDa e 5 kDa. Estes se depositam no estroma corneano, pele, parede vascular, nervos periféricos e glomérulos renais, causando os sintomas multiorgânicos característicos da síndrome de Meretoja2,11). Os depósitos corneanos frequentemente precedem outros sintomas sistêmicos, podendo ser a primeira oportunidade para o oftalmologista diagnosticar a doença.
Foram relatados casos de LCD devido a mutações de novo no gene TGFBI1). Mesmo em casos sem histórico familiar, deve-se considerar a possibilidade de mutação de novo, sendo recomendada a confirmação por teste genético1). A mutação L509P é rara, mas apresenta fenótipos variados, desde distrofia corneana de Reis-Bücklers até LCD tipo IIIA1).
No gene GSN, além das mutações clássicas p.Asp187Asn/Tyr, foi relatada uma nova mutação p.Glu580Lys, que se mostrou associada a distrofia corneana em treliça, flacidez cutânea, arritmia cardíaca, nefropatia e neuropatia óptica, caracterizando uma amiloidose sistêmica2).
Lesões corneanas posteriores e manejo cirúrgico intraocular
Foi demonstrado patologicamente que depósitos amiloides estão presentes na córnea posterior de pacientes com LCD1, podendo afetar a adesão da membrana de Descemet3). É necessário estar atento ao risco de descolamento da membrana de Descemet durante cirurgias intraoculares, como a cirurgia de catarata.
Esse achado tem implicações clínicas na avaliação da indicação e no planejamento da técnica cirúrgica para catarata em pacientes com LCD1.
Procedimentos cirúrgicos mais precisos, como a ceratectomia lamelar assistida por laser de femtosegundo (femtosecond laser-assisted lamellar keratectomy, FLK) e a ceratoplastia lamelar assistida por laser de femtosegundo (femtosecond laser-assisted lamellar keratoplasty, FALK), estão sendo desenvolvidos 12). Eles estão sendo posicionados como opções complementares à PTK convencional, devido à melhora na suavidade da superfície de corte e ao controle de profundidade altamente reprodutível.
Como a mutação TGFBI é uma mutação autossômica dominante de ganho de função, siRNA específico para o alelo mutante, oligonucleotídeos antissenso e knockout alélico específico por CRISPR-Cas9 estão sendo investigados em estágio pré-clínico. A córnea é vantajosa como órgão-alvo para terapia genética por permitir administração tópica e possuir privilégio imunológico. No entanto, atualmente não há aplicação clínica, e todas necessitam de validação futura de segurança e eficácia a longo prazo.
Inibidores da formação de amiloide e chaperonas moleculares
Compostos de baixo peso molecular que visam o processo de agregação de TGFBIp e gelsolina mutante, chaperonas moleculares (como indutores de Hsp70) e inibidores da ligação de fibrilas amiloides estão sendo investigados em estágio de pesquisa básica. Para a amiloidose tipo gelsolina sistêmica, fármacos que inibem a clivagem da gelsolina mutante no plasma estão sendo avaliados em alguns estudos pré-clínicos 2). Futuramente, espera-se que essas terapias moleculares direcionadas se tornem tratamentos curativos, substituindo a excise física convencional (PTK e transplante de córnea).
A análise proteômica da córnea por espectrometria de massas sugere que, nos depósitos da LCD1, não apenas TGFBIp, mas também múltiplas proteínas anormais podem co-precipitar. Para futura aplicação clínica, a contribuição patológica dessas proteínas co-precipitadas está sendo elucidada.
Ji YW, Ahn H, Shin KJ, Kim TI, Seo KY, Stulting RD, et al. De Novo L509P Mutation of the TGFBI Gene Associated with Slit-Lamp Findings of Lattice Corneal Dystrophy Type IIIA. Journal of clinical medicine. 2022;11(11). doi:10.3390/jcm11113055. PMID:35683443; PMCID:PMC9181583.
Potrc M, Volk M, de Rosa M, Pizem J, Teran N, Jaklic H, Maver A, Drnovsek-Olup B, Bollati M, Vogelnik K, Hocevar A, Gornik A, Pfeifer V, Peterlin B, Hawlina M, Fakin A. Clinical and Histopathological Features of Gelsolin Amyloidosis Associated with a Novel GSN Variant p.Glu580Lys. Int J Mol Sci. 2021;22(3):1084.
Matsumoto A, Fukuoka H, Sotozono C. Descemet’s Membrane Detachment During Cataract Surgery in Lattice Corneal Dystrophy Type I: Histopathological Analysis of Posterior Corneal Involvement. Cureus. 2025;17(3):e81431. doi:10.7759/cureus.81431. PMID:40296953; PMCID:PMC12037203.
Weiss JS, Møller HU, Aldave AJ, et al. IC3D classification of corneal dystrophies-Edition 2. Cornea. 2015;34(2):117-159. doi:10.1097/ICO.0000000000000307. PMID:25564336.
Han KE, Choi SI, Kim TI, Maeng YS, Stulting RD, Ji YW, et al. Pathogenesis and treatments of TGFBI corneal dystrophies. Progress in retinal and eye research. 2016;50:67-88. doi:10.1016/j.preteyeres.2015.11.002. PMID:26612778.
Lisch W, Seitz B. The clinical landmarks of corneal dystrophies. Developments in ophthalmology. 2011;48:9-23. doi:10.1159/000324075. PMID:21540629.
American Academy of Ophthalmology Cornea/External Disease Panel. Corneal Edema and Opacification Preferred Practice Pattern. San Francisco, CA: American Academy of Ophthalmology; 2024.
Dinh R, Rapuano CJ, Cohen EJ, Laibson PR. Recurrence of corneal dystrophy after excimer laser phototherapeutic keratectomy. Ophthalmology. 1999;106(8):1490-1497.
Marcon AS, Cohen EJ, Rapuano CJ, Laibson PR. Recurrence of corneal stromal dystrophies after penetrating keratoplasty. Cornea. 2003;22(1):19-21.
Meretoja J. Familial systemic paramyloidosis with lattice dystrophy of the cornea, progressive cranial neuropathy, skin changes and various internal symptoms. A previously unrecognized heritable syndrome. Ann Clin Res. 1969;1(4):314-324. PMID:4313418.
Kivelä T, Tarkkanen A, Frangione B, Ghiso J, Haltia M. Ocular amyloid deposition in familial amyloidosis, Finnish: an analysis of native and variant gelsolin in Meretoja’s syndrome. Invest Ophthalmol Vis Sci. 1994;35(10):3759-3769. PMID:8088963.
Steger B, Romano V, Biddolph S, Willoughby CE, Batterbury M, Kaye SB. Femtosecond laser-assisted lamellar keratectomy for corneal opacities secondary to anterior corneal dystrophies: an interventional case series. Cornea. 2016;35(1):6-13. PMID:26509759. doi:10.1097/ICO.0000000000000665.
Copie o texto do artigo e cole no assistente de IA de sua preferência.
Artigo copiado para a área de transferência
Abra um assistente de IA abaixo e cole o texto copiado na conversa.