حثل القرنية الشبكي (lattice corneal dystrophy, LCD) هو حثل قرنية وراثي يتميز بترسب الأميلويد في سدى القرنية مسببًا عتامة خطية شبكية. تم وصفه لأول مرة في تسعينيات القرن التاسع عشر، ويُصنف حاليًا في التصنيف الدولي لحثل القرنية (IC3D) الإصدار الثاني إلى LCD1 ومتغيراته (الأنواع 3، 3A، 1/3A، 4 سابقًا)4).
يشكل LCD1 وحثل القرنية الحبيبي وحثل ريس-بوكلرز وحثل ثيل-بينكه مجموعة من الأمراض تُعرف باسم “حثل القرنية المرتبط بـ TGFBI”. يقع الجين المسبب TGFBI (جين محفز بعامل النمو المحول بيتا) على الذراع الطويل للكروموسوم 5 (5q31)، ويتبع نمط الوراثة الجسدية السائدة. يُنتج بروتين TGFBI (TGFBIp، كيراتو-إبيثيلين، βig-h3) بواسطة ظهارة القرنية ويتوزع في جميع طبقات القرنية. في سدى القرنية، يشارك في بناء ألياف الكولاجين. حتى مع وجود طفرات في نفس الجين، يؤدي اختلاف موقع الطفرة ونوع الحمض الأميني المستبدل إلى تمايز كبير في المادة المترسبة (هيالين أم أميلويد) والصورة السريرية5).
الطفرة الممثلة لـ LCD1 هي R124C، حيث يتم استبدال الأرجينين في الموضع 124 من جين TGFBI بالسيستين. في النوع المتغير LCD IIIA، تم الإبلاغ عن طفرات مثل L527R.
البروتين غير الطبيعي المتراكم في القرنية يظهر باللون الأحمر عند صبغه بالكونغو الأحمر، ويظهر انكسارًا مزدوجًا أخضر تفاحيًا مميزًا تحت المجهر المستقطب، مما يؤكد أنه أميلويد. هذا هو المؤشر التشخيصي النسيجي الكلاسيكي للداء النشواني منذ القرن التاسع عشر6).
النوع الذي كان يُسمى سابقًا “الحثل الشبكي القرني من النوع 2” هو عرض عيني للداء النشواني الجهازي من نوع جيلسولين (GSN-AMYL، متلازمة ميريتويا)، ويُصنف حاليًا ضمن “الداء النشواني العائلي” في تصنيف IC3D، ويُعالج بشكل مستقل عن LCD الكلاسيكي4,10). تم وصف هذه المتلازمة لأول مرة بواسطة ميريتويا في فنلندا عام 1969، وهي مرض وراثي يتميز بعتامة شبكية قرنية بالإضافة إلى اعتلال عصبي قحفي تقدمي وترهل الجلد وأعراض جهازية10,11). نظرًا لأهمية التمييز بينهما في الممارسة السريرية، يتم تضمين كليهما في هذه المقالة.
عتامات خيطية مزدوجة المحيط في منطقة الحدقة، تآكل ظهاري متكرر
LCD IIIA (نوع متغير)
TGFBI (5q31)
L527R وغيره
بعد سن الأربعين
خطوط شبكية سميكة تشبه الحبل في عمق السدى، لا تآكل ظهاري
نوع GSN (ميريتويا)
GSN (9q34)
D187N، p.Glu580Lys2)
30-40 سنة
خطوط شبكية شعاعية في المحيط، داء نشواني جهازي
في اليابان، الحثل المرتبط بـ TGFBI الأكثر شيوعًا هو النوع الحبيبي II (نوع أفيلينو، R124H)، وLCD1 أقل شيوعًا مقارنة به. ومع ذلك، نظرًا لأن كليهما ينشأان من اختلاف بضع قواعد نيتروجينية فقط في نفس جين TGFBI، فمن المستحسن تأكيد التشخيص بالاختبار الجيني في الحالات التي تتداخل فيها الصور السريرية. لم يتم الإبلاغ عن معدل الانتشار الدقيق لـ LCD ككل في اليابان، لكنه يعتبر نادرًا نسبيًا بين حالات حثل القرنية.
Qما الفرق بين LCD1 ومتلازمة ميريتويا؟
A
LCD1 هو ترسب نشواني موضعي في القرنية ناتج عن طفرة في جين TGFBI، ويبدأ في منطقة الحدقة في العقد الثاني أو الثالث من العمر، ويصاحبه تآكل ظهاري متكرر بشكل متكرر. في المقابل، متلازمة ميريتويا (LCD2 سابقًا، نوع GSN) هي مظهر عيني لداء النشواني الجهازي الناتج عن طفرة في جين GSN (جيلسولين)، وتبدأ في محيط القرنية في العقد الثالث أو الرابع من العمر، وتحافظ شفافية المركز لفترة طويلة. في متلازمة ميريتويا، توجد أعراض جهازية مثل ترهل الجلد، وجه يشبه القناع، واعتلال الأعصاب المحيطية، وعدم انتظام ضربات القلب2,10). في الإصدار الثاني من IC3D، تُصنف متلازمة ميريتويا بشكل مستقل عن حثل القرنية الشبكي4).
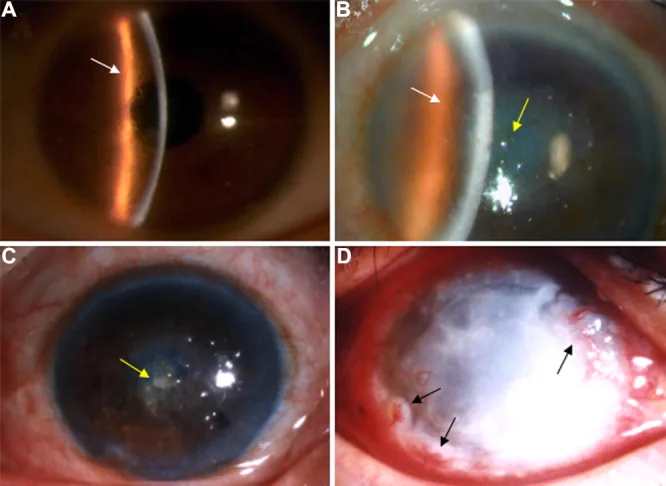
Liu Z, et al. An R124C mutation in TGFBI caused lattice corneal dystrophy type I with a variable phenotype in three Chinese families. Mol Vis. 2008. Figure 2. PMCID: PMC2443752. License: CC BY.
في صورة المصباح الشقي، تظهر خطوط شبكية متفرعة في سدى القرنية وعتامة مركزية سائدة. هذه صورة تمثل النتائج السريرية النموذجية لحثل القرنية الشبكي.
في LCD1، يكون معظم المرضى بدون أعراض في مرحلة الطفولة، ولا تظهر سوى عتامة دقيقة يمكن اكتشافها فقط باستخدام الإضاءة الخلفية للمصباح الشقي. بعد العقد الثاني أو الثالث من العمر، تحدث تآكلات ظهارية قرنية متكررة (RCE) بشكل متكرر، مع ألم حاد في العين عند الاستيقاظ، رهاب الضوء، الدمع، والإحساس بجسم غريب. حوالي سن الثلاثين، تظهر عتامة بيضاء في الطبقات السطحية من سدى القرنية المركزية، ويتدهور البصر بعد سن الأربعين.
في LCD IIIA (النوع المتغير)، لا يحدث عادةً ضرر ظهاري، ويكون الشكوى الرئيسية هي تدهور البصر التدريجي بعد سن الأربعين.
في LCD2 القديم (متلازمة ميريتويا)، تظهر الأعراض العينية في الثلاثينيات والأربعينيات من العمر، ولكن غالبًا ما يتأخر ضعف البصر الشديد حتى الستينيات11). غالبًا ما تسبق أو تصاحب الأعراض الجهازية مثل ارتخاء جلد الجفن، الوجه القناعي، الاعتلال العصبي القحفي التدريجي، وعدم انتظام ضربات القلب2,10).
موقع البداية: تظهر كعتامات دقيقة نقطية أو خطية في منطقة الحدقة في كلتا العينين، في طبقة بومان إلى السدى السطحي.
الخطوط الشبكية: عتامات خيطية أو خطية ذات حدود مزدوجة تتشابك لتشكل عتامة شبكية أو نجمية.
المرحلة المتقدمة: تظهر عتامة بيضاء حليبية بيضاوية أو دائرية في القرنية المركزية.
الإضاءة الخلفية: تظهر الخطوط الشبكية الرقيقة الشفافة التي يصعب رؤيتها بالإضاءة المباشرة بوضوح.
صبغة الفلوريسئين: بسبب ضعف التصاق الظهارة، يصبح سطح القرنية خشنًا.
التآكل الظهاري المتكرر: يحدث بشكل متكرر لأن الرواسب تمتد إلى الخلايا القاعدية الظهارية وغشاء بومان.
LCD IIIA (النوع المتغير)
خطوط شبكية: خطوط شبكية سميكة وطويلة في الطبقة الوسطى إلى العميقة من السدى، تظهر أحيانًا بتفرعات شجرية. يمكن ملاحظتها حتى بالإضاءة المباشرة.
النمط الظاهري: هناك ثلاثة أنماط: ① خطوط شبكية فقط، ② رواسب حبيبية صغيرة فقط، ③ مزيج من الاثنين. قد تظهر العين اليمنى واليسرى نمطًا ظاهريًا مختلفًا لدى نفس الفرد، وقد تكون الحالة أحادية العين.
الظهارة: عادةً لا تحدث اضطرابات ظهارية.
متماثل الزيجوت: في متماثلي الزيجوت L527R، تكون الخطوط الشبكية أكثر سمكًا والرواسب الحبيبية المركزية أكبر، لكن الفرق ليس واضحًا كما هو بين متغاير ومتماثل الزيجوت R124H (النوع الحبيبي II).
نوع GSN (ميريتويا)
خطوط شبكية: عدد قليل من الرواسب الشبكية غير الدقيقة تظهر بشكل شعاعي من المحيط.
شفافية مركزية: تبقى المنطقة المركزية شفافة لفترة طويلة بعد ظهور المرض.
تآكل ظهاري: نادر.
العلامات الجهازية: تغيرات في الوجه مثل الوجه القناعي، شفاه بارزة مع اضطرابات حركية، آذان متدلية، وترهل جلد الجفن2).
في LCD1، قد تكون العتامة المركزية الدائرية شديدة في بعض الحالات؛ فقد تم الإبلاغ عن حالة لمريض يبلغ من العمر 56 عامًا متغاير الزيجوت R124C خضع لزراعة القرنية بسبب عتامة مركزية دائرية.
Qهل يمكن تشخيص LCD1 لدى الأطفال؟
A
معظم حالات LCD1 في مرحلة الطفولة تكون بدون أعراض، ويصعب اكتشاف التشوهات بالإضاءة المباشرة فقط. يمكن ملاحظة عتامات دقيقة نقطية إلى خطية في الطبقة السطحية من السدى المركزي باستخدام الفحص التفصيلي بمنظار الشق مع الإضاءة الخلفية أو الإضاءة المرتدة. يُوصى بمراعاة LCD1 لدى الأطفال الذين يعانون من تآكل ظهاري متكرر، مع أخذ التاريخ العائلي وفحص قرنية الوالدين. يُعد اختبار جين TGFBI مفيدًا للتشخيص النهائي.
فيما يلي ملخص للجينات المسببة والطفرات النموذجية لحثل القرنية الشبكي.
مرتبط بـ TGFBI (LCD1، LCD IIIA، LCD IV)
الموضع الجيني: 5q31 (جين TGFBI).
نمط الوراثة: وراثة جسمية سائدة.
الطفرة الأكثر شيوعًا في LCD1: R124C (Arg124Cys) هي الأكثر تكرارًا5).
الطفرات الممثلة لـ LCD IIIA: تم الإبلاغ عن طفرات مثل L527R (Leu527Arg). توجد أيضًا حالات متماثلة اللواقح.
الطفرات الجديدة: تم الإبلاغ عن حالة لطفرة جديدة L509P في جين TGFBI أظهرت النمط الظاهري لـ LCD IIIA1). لم تكن الطفرة موجودة لدى الوالدين، وورثها أحد الأبناء1).
دور TGFBIp: تنتجه ظهارة القرنية ويتوزع في جميع طبقات القرنية، ويشارك في بناء ألياف الكولاجين في السدى5).
المرتبط بـ GSN (متلازمة ميريتويا، سابقًا LCD2)
الموضع الجيني: 9q34 (جين GSN، جيلسولين).
نمط الوراثة: وراثة جسمية سائدة.
الطفرة التقليدية: D187N (النمط الفنلندي) هي الأكثر شيوعًا، كما تم الإبلاغ عن p.Asp187Tyr10,11).
طفرة جديدة: تم الإبلاغ عن طفرة p.Glu580Lys في عائلة سلوفينية تقع على حدود المجالين G4-G5، وتسبب تنافرًا كهروستاتيكيًا بسبب استبدال الشحنة السالبة بشحنة موجبة2).
الصورة السريرية: بالإضافة إلى عتامة القرنية الشبكية، تظهر أعراض الداء النشواني الجهازي المصحوب بترهل الجلد، وعدم انتظام ضربات القلب، واعتلال الكلى، واعتلال العصب البصري2).
نظرًا لأنه مرض وراثي، فإن التاريخ العائلي هو أهم عامل خطر. ومع ذلك، يمكن أن تحدث طفرات جديدة في جين TGFBI، لذا لا يمكن استبعاد المرض بغياب التاريخ العائلي1). نمط الوراثة هو جسمي سائد، وإذا كان أحد الوالدين حاملًا للطفرة، فإن احتمال انتقالها إلى الطفل هو 50%. لا يوجد فرق بين الجنسين، ولا يوجد اختلاف عرقي واضح في LCD1، لكن متلازمة ميريتويا معروفة بتراكم العائلات في فنلندا11).
ليس من الواضح مساهمة العوامل البيئية، ويتم تحديد بداية المرض وتطوره بشكل أساسي من خلال النمط الجيني. ومع ذلك، قد يزداد تواتر تآكل الظهارة المتكرر في البيئات الجافة، أو استخدام العدسات اللاصقة، أو الصدمات. يمكن أن تسبب جراحات تصحيح البصر (مثل LASIK و SMILE) تفاقمًا سريعًا لحثل القرنية المرتبط بـ TGFBI، لذا يجب توخي الحذر في الحالات التي لديها تاريخ عائلي أثناء الفحص قبل الجراحة5).
لتشخيص LCD1 والنوع المتغير ونوع GSN، يتم الجمع بين نتائج المصباح الشقي والأنسجة والجينات.
الفحوصات السريرية
مصباح الشق المجهري: في الإضاءة المباشرة، من السهل تفويت الخطوط الشبكية المبكرة. باستخدام طريقة الإضاءة الخلفية، يتم الكشف عن العتامات الدقيقة على خلفية الحدقة، وباستخدام الإضاءة المرتدة، يتم الكشف عن الخطوط الشبكية الرقيقة الشفافة.
صبغة الفلوريسئين: في LCD1، يصبح التصاق الظهارة ضعيفًا، مما يجعل الصبغة تبدو خشنة. كما أنها مفيدة لتقييم مدى التآكل الظهاري.
التصوير المقطعي للجزء الأمامي (OCT): يمكن قياس عمق الترسبات طبقيًا. قياس عمق الآفة باستخدام FD-OCT مفيد لتحديد عمق الاستئصال في PTK1).
المجهر البؤري للقرنية: يمكن ملاحظة الترسبات داخل السدى على المستوى الخلوي.
التشخيص النهائي
الاختبار الجيني: يتم تأكيد النوع المرضي عن طريق الكشف عن طفرات في جينات TGFBI و GSN. حتى مع نفس النمط الظاهري، تختلف سرعة التكرار والتقدم باختلاف الطفرة، مما يؤثر مباشرة على خطة العلاج.
الفحص المرضي: يظهر اللون الأحمر بصبغة الكونغو الأحمر، ويظهر انكسارًا مزدوجًا أخضر تفاحي تحت المجهر المستقطب، مما يؤكد وجود الأميلويد6).
الكيمياء المناعية النسيجية: يمكن التمييز بين الأنواع المرضية باستخدام الأجسام المضادة لـ TGFBIp والأجسام المضادة للجيلسولين.
أخذ التاريخ العائلي: نظرًا لأن الوراثة هي صبغية جسدية سائدة، فإن فحص قرنية الوالدين والأشقاء يدعم التشخيص.
حثل القرنية الحبيبي النوع الثاني (نوع أفيلينو، TGFBI R124H): هو أكثر أنواع حثل القرنية المرتبط بـ TGFBI شيوعًا في اليابان، ويظهر مزيجًا من الترسبات الحبيبية والخطوط الشبكية. لتشخيصه التفريقي عن LCD1، يكون الاختبار الجيني موثوقًا.
الداء النشواني القرني الثانوي: ليس وراثيًا، ويترسب النشواني بشكل ثانوي نتيجة لتحفيز مزمن لسطح العين مثل الرموش المنحرفة أو القرنية المخروطية. يتميز بعدم وجود تاريخ عائلي ووجود مرض أساسي.
حثل القرنية البقعي: وراثة جسمية متنحية ناتجة عن طفرة في جين CHST6، ويصاحبه عتامة منتشرة تشبه الزجاج المصنفر وشذوذ في البطانة.
حثل القرنية القطروي الغرواني: وراثة جسمية متنحية ناتجة عن طفرة في جين TACSTD2، ويظهر على شكل نتوءات غروانية بيضاء حليبية. شائع نسبيًا في اليابان.
Qلماذا يعتبر الفحص الجيني مهمًا؟
A
في حثل القرنية الشبكي، حتى لو كانت الأنماط الظاهرية متشابهة، فإن اختلاف الجين المسبب وموقع الطفرة يؤدي إلى اختلاف كبير في سرعة التقدم، وتكرار الانتكاس، وخيارات العلاج، ووجود مضاعفات جهازية. يختلف LCD1 الناتج عن طفرة TGFBI ومتلازمة ميريتويا الناتجة عن طفرة GSN اختلافًا جوهريًا في خطة العلاج والحاجة إلى الفحص الجهازي2,10). بالإضافة إلى ذلك، تم الإبلاغ عن حالات ذات طفرات جديدة (de novo) لا يمكن تحديد نوع المرض فيها بالاعتماد على التاريخ العائلي فقط1)، مما يجعل الفحص الجيني ضروريًا للتشخيص النهائي وتصنيف النوع.
في مرحلة الطفولة إلى الشباب، عندما لا تظهر أعراض أو يكون هناك عتامة طفيفة فقط، يتم إجراء المراقبة. يتم تقييم التقدم عن طريق فحص المصباح الشقي كل 6 أشهر إلى سنة.
بالنسبة لتآكل القرنية المتكرر، وهو أحد الأعراض الأساسية لـ LCD1، فإن العلاج التحفظي التالي هو الخطوة الأولى:
علاج النوبات: استخدام العدسات اللاصقة اللينة العلاجية بشكل مستمر لحماية ظهارة القرنية. يُستخدم معها قطرات مضادة للبكتيريا للوقاية من العدوى الثانوية. يُستخدم مرهم للعين للتزليق وحماية الظهارة.
الوقاية من الانتكاس: يساعد استخدام مرهم العين قبل النوم في تقليل تكرار نوبات تآكل القرنية المتكرر. في البيئات الجافة، يُستخدم الدموع الاصطناعية أو مواد التزليق أثناء النهار أيضًا.
في LCD1، حيث يكون ترسب النشواني في الطبقة السطحية للقرنية هو السائد، وفي حالات العتامة المركزية الشديدة أو تآكل القرنية المتكرر، يُعد استئصال القرنية الضوئي العلاجي (PTK) باستخدام ليزر الإكسيمر الخيار الأول7,8). عادةً لا يحدث انتكاس مبكر، لكن الانتكاس مع مرور الوقت لا مفر منه، ويمكن إجراء PTK على نفس العين حتى مرتين.
في الحالات متغايرة الزيجوت، يكون التكرار بطيئًا ونادرًا ما يحتاج إلى علاج متكرر. في الحالات متماثلة الزيجوت، يميل التكرار إلى الحدوث مبكرًا مقارنة بالحالات متغايرة الزيجوت. يزداد معدل التكرار بعد PTK مع مرور الوقت، كما هو الحال في حالات ضمور TGFBI الأخرى، وتظهر علامات التكرار في معظم الحالات عند المتابعة طويلة المدى8).
كمثال على فعالية PTK، في حالة LCD IIIA ناتجة عن طفرة de novo في TGFBI L509P، تم إجراء PTK بعمق 60 ميكرومتر تحت توجيه FD-OCT، وتحسنت أفضل حدة بصرية مصححة (BCVA) من 20/400 إلى 20/501). لم يلاحظ انخفاض في حدة البصر أو تكرار كبير بعد 45 شهرًا من الجراحة1).
وفقًا لنمط الممارسة المفضل لـ AAO لعتامة ووذمة القرنية، فإن PTK لضمور القرنية الحبيبي والشبكي هو “خيار معقول” وقد يؤخر الانتقال إلى DALK أو زرع القرنية كامل السمك، ولكنه يحمل خطر حدوث عتامة بعد الجراحة. عند التكرار، يُنظر في استخدام الميتوميسين C معًا كوسيلة لكبح الندبات المتكررة أو الترسبات اللحمية، ويُحذر من زيادة خطر توسع القرنية إذا تجاوز الاستئصال الثلث الأمامي من اللحمة أو إذا كان السرير المتبقي أقل من 250 ميكرومتر7).
في حالات التكرار المتكرر أو عندما تمتد العتامة إلى طبقات أعمق من اللحمة، يتم اختيار زرع القرنية. في LCD1، عادة لا يكون زرع القرنية ضروريًا حتى سن الأربعين. نظرًا لأن الخلايا البطانية للقرنية طبيعية في LCD، يتم اختيار التقنية الجراحية بناءً على عمق العتامة.
في السنوات الأخيرة، أصبحت عملية رأب القرنية العميقة (DALK) الخيار الأول الجديد المستخدم على نطاق واسع نظرًا لانخفاض خطر الرفض وتحقيق نتائج بصرية مماثلة لرأب القرنية كامل السمك.
تكرار مرض الحثل اللاتي القرني (LCD) بعد زرع القرنية أمر لا مفر منه، وقد تم الإبلاغ عن معدلات التكرار بعد زرع القرنية كامل السمك بنسبة 17.8% في 5 سنوات، و26% في 8 سنوات، و56% في 15 عامًا9). نظرًا لأن العتامة المتكررة تقتصر عادةً على الطبقة السطحية، يمكن إزالتها باستخدام استئصال القرنية الضوئي العلاجي (PTK) مما يطيل الفترة حتى إعادة الزرع. بالنسبة لـ LCD IIIA (النوع المتغير)، غالبًا لا يتطلب العلاج ما لم يكن التأثير على الرؤية شديدًا.
Qما مدى فعالية PTK؟
A
يمكن لـ PTK إزالة ترسبات الأميلويد السطحية بشكل فعال، مما يحسن الرؤية ويقلل من تآكل الظهارة المتكرر. في حالة LCD IIIA، تم الإبلاغ عن تحسن حدة البصر المصححة من 20/400 إلى 20/50 بعد PTK بعمق 60 ميكرومتر، مع عدم حدوث تكرار لمدة 45 شهرًا1). في متغايري الزيجوت، يكون التكرار بطيئًا، بينما في متماثلي الزيجوت يحدث تكرار مبكر. لا يمكن إزالة الآفات العميقة باستخدام PTK، لذا تتطلب العتامة العميقة إجراء DALK أو زرع قرنية كامل السمك7).
Qهل يتكرر المرض بعد زرع القرنية؟
A
تكرار LCD بعد زرع القرنية أمر لا مفر منه. تم الإبلاغ عن معدلات التكرار بعد زرع القرنية كامل السمك بنسبة 17.8% في 5 سنوات، و26% في 8 سنوات، و56% في 15 عامًا9). ومع ذلك، فإن العتامة المتكررة تقتصر عادةً على الطبقة السطحية للطعم، لذا يمكن إزالتها باستخدام PTK مما يطيل عمر الطعم. تتميز عملية رأب القرنية العميقة (DALK) بانخفاض خطر رفض الطبقة البطانية مقارنة بزرع القرنية كامل السمك، وتعتبر الخيار الأول الجديد7).
يتمثل جوهر مرض LCD1 في التراكم غير الطبيعي لبروتين TGFBIp (kerato-epithelin، βig-h3). يُنتج TGFBIp عادةً بواسطة ظهارة القرنية ويتوزع في جميع طبقات القرنية، ويشارك في بنية ألياف الكولاجين والتصاق الخلايا في السدى 5). يؤدي البروتين غير الطبيعي الناتج عن طفرة R124C إلى سوء الطي والتجمع الذاتي، ويترسب كألياف أميلويد غير قابلة للذوبان في طبقة بومان والطبقات السطحية من السدى. في المراحل المتقدمة، ينتشر الترسيب إلى الطبقات العميقة من السدى.
يسبب ترسب الأميلويد تغيرات في بنية التصاق الظهارة الأمامية للقرنية، مما يؤدي إلى تنكس الخلايا القاعدية الظهارية وتنكس الطبقة الظهارية مصحوبًا بفقدان غشاء بومان. يشكل هذا الانهيار البنيوي الأساس المرضي لتآكلات الظهارة المتكررة.
في جين TGFBI، يحدد موقع الطفرة والحمض الأميني البديل الصورة السريرية. تسبب R124C مرض LCD1، وتسبب R124H الحثل القرني الحبيبي من النوع الثاني (نوع أفيلينو)، وتسبب R124L حثل قرنية رايس-بوكلرز 5). الآلية الجزيئية التي يحدد بها اختلاف حمض أميني واحد مادة الترسيب (أميلويد مقابل هيالين مقابل كليهما) وموقع الترسيب لم تُفهم بالكامل بعد، لكن يُعتقد أن موقع الطفرة في أي مجال من مجالات βig-h3 وتأثيرها على استقرار الطي هما المفتاح.
في LCD IIIA، تسبب طفرات سائدة في الطبقات العميقة مثل L527R خطوطًا شبكية سميكة تشبه الحبل، وتكون متأخرة الظهور دون إصابة ظهارية. يمكن تفسير التوطين الطبقي للترسبات من خلال تدرج إفراز وانتشار βig-h3 من الخلايا المنتجة (الخلايا القاعدية الظهارية) إلى السدى، واختلاف استقرار طي البروتين الطافر. يُعتقد أن R124C يفضل مسارًا من وسيط الطي إلى تكوين ألياف الأميلويد، مما يترسب الأميلويد حول طبقة بومان5). بينما تشكل طفرة L527R بروتينًا مطويًا بشكل خاطئ مستقرًا نسبيًا، ويترسب ببطء في الطبقات العميقة من السدى.
تقليديًا، كان يُعتقد أن ترسب الأميلويد في LCD1 يقتصر على القرنية الأمامية (طبقة بومان والطبقات السطحية من السدى). لكن الدراسات المرضية الحديثة أظهرت وجود ترسب أميلويد في القرنية الخلفية بالقرب من غشاء ديسيميه 3). قد يؤثر ترسب الأميلويد في القرنية الخلفية على التصاق غشاء ديسيميه، مما يساهم في انفصال غشاء ديسيميه أثناء جراحة الساد 3). يُقترح أن نفس الآلية التي يضعف بها ترسب الأميلويد التصاق الظهارة في القرنية الأمامية تعمل أيضًا في الجزء الخلفي 3).
جيلسولين، الجزيء المسبب لمتلازمة ميريتويا (LCD2 سابقًا)، يوجد في كل من السيتوبلازم وخارج الخلية، وهو بروتين يشارك في حركة الخلية وانقسامها وموتها المبرمج عبر ارتباطه بالأكتين. الطفرة الكلاسيكية D187N، المعروفة بالنمط الفنلندي، تسبب ترسبات شبكية في القرنية واعتلالًا عصبيًا قحفيًا كأعراض رئيسية11). الطفرة الجديدة p.Glu580Lys التي تم الإبلاغ عنها في عائلة سلوفينية تقع على حدود المجالين G4-G5، وتؤدي إلى استبدال حمض الجلوتاميك سالب الشحنة بالليسين موجب الشحنة، مما يسبب تنافرًا كهروستاتيكيًا ويقلل من الترابط والاستقرار بين المجالات2). يخضع الجيلسولين المتحور لانشقاق غير طبيعي بواسطة فورين وMT1-MMP في البلازما، مما يطلق شظايا أولية من الأميلويد بحجم 8 كيلو دالتون و5 كيلو دالتون. تترسب هذه الشظايا في سدى القرنية والجلد وجدران الأوعية الدموية والأعصاب المحيطية وكبيبات الكلى، مما يسبب أعراضًا متعددة الأعضاء مميزة لمتلازمة ميريتويا2,11). غالبًا ما تسبق الترسبات القرنية الأعراض الجهازية الأخرى، مما قد يجعل طبيب العيون أول من يشخص هذا المرض.
تم الإبلاغ عن حدوث حثل القرنية الشبكي (LCD) بسبب طفرات جديدة (de novo) في جين TGFBI1). حتى في الحالات التي لا يوجد فيها تاريخ عائلي، يجب مراعاة احتمال وجود طفرة جديدة، ويوصى بالتأكيد عبر الفحص الجيني1). الطفرة L509P نادرة ولكنها تسبب أنماطًا ظاهرية متنوعة تتراوح بين حثل رايس-بكلرز الشبيه بحثل LCD IIIA1).
في جين GSN، بالإضافة إلى الطفرات التقليدية p.Asp187Asn/Tyr، تم الإبلاغ عن طفرة جديدة p.Glu580Lys، والتي تسبب داء نشواني جهازي مصحوب بحثل القرنية الشبكي وترهل الجلد وعدم انتظام ضربات القلب واعتلال الكلى واعتلال العصب البصري2).
أظهرت الدراسات المرضية وجود ترسبات أميلويد في القرنية الخلفية لمرضى LCD1، مما قد يؤثر على التصاق غشاء ديسيميه3). يجب الانتباه إلى خطر انفصال غشاء ديسيميه أثناء الجراحة داخل العين، مثل جراحة الساد.
لهذه النتيجة آثار سريرية على تقييم مؤشرات جراحة الساد وتخطيط التقنية الجراحية لمرضى LCD1.
يجري تطوير إجراءات جراحية أكثر دقة مثل استئصال القرنية الرقائقي بمساعدة ليزر الفيمتو ثانية (FLK) وزرع القرنية الرقائقي بمساعدة ليزر الفيمتو ثانية (FALK) 12). تُعتبر هذه الإجراءات خيارات مكملة لـ PTK التقليدية بفضل تحسين نعومة سطح القطع والتحكم الدقيق في العمق.
نظرًا لأن طفرة TGFBI هي طفرة سائدة جسمية مكتسبة الوظيفة، يتم دراسة الحمض النووي الريبي المتداخل الخاص بالأليل المتغير (siRNA) والأليغنوكليوتيدات المضادة للاتجاه (antisense oligonucleotides) والاستهداف الجيني الخاص بالأليل باستخدام CRISPR-Cas9 في مرحلة الدراسات قبل السريرية. القرنية هي عضو مستهدف مناسب للعلاج الجيني لأنها قابلة للتطبيق الموضعي ولها امتياز مناعي. ومع ذلك، لا يوجد حاليًا أي تطبيق سريري، وكلها تتطلب التحقق من السلامة والفعالية على المدى الطويل.
يتم دراسة المركبات الجزيئية الصغيرة التي تستهدف عملية تجمع TGFBIp والجيلسولين المتحور، والمرافقات الجزيئية (مثل محفزات Hsp70)، ومثبطات ارتباط ألياف الأميلويد في مرحلة الأبحاث الأساسية. بالنسبة للداء النشواني من نوع الجيلسولين الجهازي، يتم تقييم الأدوية التي تثبط مرحلة قطع الجيلسولين المتحور في البلازما في بعض التجارب قبل السريرية 2). من المتوقع أن تحل هذه العلاجات الجزيئية المستهدفة محل الاستئصال الجراحي التقليدي (PTK وزرع القرنية) كعلاج جذري في المستقبل.
أظهر تحليل بروتيوم القرنية باستخدام مطياف الكتلة أن رواسب LCD1 قد تحتوي على ترسب مشترك لبروتينات غير طبيعية متعددة بالإضافة إلى TGFBIp. يتم دراسة مساهمة هذه البروتينات المترسبة بشكل مشترك في المرض من أجل التطبيق السريري المستقبلي.
Ji YW, Ahn H, Shin KJ, Kim TI, Seo KY, Stulting RD, et al. De Novo L509P Mutation of the TGFBI Gene Associated with Slit-Lamp Findings of Lattice Corneal Dystrophy Type IIIA. Journal of clinical medicine. 2022;11(11). doi:10.3390/jcm11113055. PMID:35683443; PMCID:PMC9181583.
Potrc M, Volk M, de Rosa M, Pizem J, Teran N, Jaklic H, Maver A, Drnovsek-Olup B, Bollati M, Vogelnik K, Hocevar A, Gornik A, Pfeifer V, Peterlin B, Hawlina M, Fakin A. Clinical and Histopathological Features of Gelsolin Amyloidosis Associated with a Novel GSN Variant p.Glu580Lys. Int J Mol Sci. 2021;22(3):1084.
Matsumoto A, Fukuoka H, Sotozono C. Descemet’s Membrane Detachment During Cataract Surgery in Lattice Corneal Dystrophy Type I: Histopathological Analysis of Posterior Corneal Involvement. Cureus. 2025;17(3):e81431. doi:10.7759/cureus.81431. PMID:40296953; PMCID:PMC12037203.
Weiss JS, Møller HU, Aldave AJ, et al. IC3D classification of corneal dystrophies-Edition 2. Cornea. 2015;34(2):117-159. doi:10.1097/ICO.0000000000000307. PMID:25564336.
Han KE, Choi SI, Kim TI, Maeng YS, Stulting RD, Ji YW, et al. Pathogenesis and treatments of TGFBI corneal dystrophies. Progress in retinal and eye research. 2016;50:67-88. doi:10.1016/j.preteyeres.2015.11.002. PMID:26612778.
Lisch W, Seitz B. The clinical landmarks of corneal dystrophies. Developments in ophthalmology. 2011;48:9-23. doi:10.1159/000324075. PMID:21540629.
American Academy of Ophthalmology Cornea/External Disease Panel. Corneal Edema and Opacification Preferred Practice Pattern. San Francisco, CA: American Academy of Ophthalmology; 2024.
Dinh R, Rapuano CJ, Cohen EJ, Laibson PR. Recurrence of corneal dystrophy after excimer laser phototherapeutic keratectomy. Ophthalmology. 1999;106(8):1490-1497.
Marcon AS, Cohen EJ, Rapuano CJ, Laibson PR. Recurrence of corneal stromal dystrophies after penetrating keratoplasty. Cornea. 2003;22(1):19-21.
Meretoja J. Familial systemic paramyloidosis with lattice dystrophy of the cornea, progressive cranial neuropathy, skin changes and various internal symptoms. A previously unrecognized heritable syndrome. Ann Clin Res. 1969;1(4):314-324. PMID:4313418.
Kivelä T, Tarkkanen A, Frangione B, Ghiso J, Haltia M. Ocular amyloid deposition in familial amyloidosis, Finnish: an analysis of native and variant gelsolin in Meretoja’s syndrome. Invest Ophthalmol Vis Sci. 1994;35(10):3759-3769. PMID:8088963.
Steger B, Romano V, Biddolph S, Willoughby CE, Batterbury M, Kaye SB. Femtosecond laser-assisted lamellar keratectomy for corneal opacities secondary to anterior corneal dystrophies: an interventional case series. Cornea. 2016;35(1):6-13. PMID:26509759. doi:10.1097/ICO.0000000000000665.
انسخ نص المقال والصقه في مساعد الذكاء الاصطناعي الذي تفضله.
تم نسخ المقال إلى الحافظة
افتح أحد مساعدي الذكاء الاصطناعي أدناه والصق النص المنسوخ في مربع المحادثة.