Решетчатая дистрофия роговицы (lattice corneal dystrophy, LCD) — это наследственная дистрофия роговицы, при которой амилоид откладывается в строме роговицы, образуя решетчатые линейные помутнения. Это заболевание с давней историей, описанное еще в 1890-х годах. Согласно клинико-генетической классификации второй версии IC3D (International Committee for Classification of Corneal Dystrophies), оно объединено в LCD1 и его варианты (ранее типы 3, 3A, 1/3A, 4)4).
LCD1, гранулярная дистрофия роговицы, дистрофия Рейса-Бюклерса и дистрофия Тиля-Бенке образуют группу «TGFBI-ассоциированных дистрофий». Ген-возбудитель TGFBI (transforming growth factor beta-induced gene) расположен на длинном плече 5-й хромосомы (5q31) и наследуется по аутосомно-доминантному типу. Белок TGFBI (TGFBIp, kerato-epithelin, βig-h3) вырабатывается эпителием роговицы и распределяется по всей ее толщине. В строме роговицы он участвует в построении коллагеновых волокон. Даже при мутациях одного и того же гена различия в месте мутации и замещающей аминокислоте приводят к значительным различиям в откладываемом веществе (гиалин или амилоид) и клинической картине5).
Наиболее частой мутацией LCD1 является R124C, при которой 124-й аргинин в гене TGFBI заменяется на цистеин. При варианте LCD IIIA сообщается о мутациях, таких как L527R.
Гистологическая диагностика и классические признаки
Аномальный белок, накапливающийся в роговице, окрашивается в красный цвет при окрашивании конго красным и под поляризационным микроскопом демонстрирует характерное яблочно-зеленое двойное лучепреломление, что подтверждает амилоид. Этот признак является классическим гистологическим критерием амилоидоза с XIX века6).
Тип, ранее называвшийся «решетчатой дистрофией роговицы 2 типа», является глазным проявлением системного гельзолинового амилоидоза (GSN-AMYL, синдром Меретойя). Согласно текущей классификации IC3D, он относится к «семейному амилоидозу» и рассматривается отдельно от классической LCD4,10). Этот синдром, описанный в 1969 году финским ученым Меретойя, представляет собой наследственное заболевание, характеризующееся решетчатым помутнением роговицы, прогрессирующей черепно-мозговой нейропатией, дряблостью кожи и системными симптомами10,11). Поскольку в клинической практике важно различать эти два состояния, в данной статье они приводятся вместе.
Двухконтурные нитевидные помутнения в зрачковой области, рецидивирующие эрозии эпителия
LCD IIIA (вариантный тип)
TGFBI (5q31)
L527R и др.
После 40 лет
Толстые веревкообразные решетчатые линии в глубоких слоях стромы, без поражения эпителия
GSN-тип (Meretoja)
GSN (9q34)
D187N, p.Glu580Lys2)
30–40 лет
Радиальные решетчатые линии на периферии, системный амилоидоз
В Японии наиболее частой TGFBI-ассоциированной дистрофией является гранулярная дистрофия II типа (тип Авеллино, R124H), а LCD1 встречается реже. Однако, поскольку оба типа различаются всего несколькими нуклеотидами в одном и том же гене TGFBI, в случаях с перекрывающейся клинической картиной желательно подтверждение с помощью генетического тестирования. Точная распространенность LCD в Японии не сообщается, но в целом среди всех дистрофий роговицы этот тип встречается относительно редко.
QВ чем разница между LCD1 и синдромом Меретойя?
A
LCD1 — это локальное отложение амилоида в роговице, вызванное мутацией гена TGFBI, которое начинается в зрачковой зоне в возрасте 10–20 лет и часто сопровождается рецидивирующей эрозией эпителия. Синдром Меретойя (ранее LCD2, GSN-тип) — это глазное проявление системного амилоидоза, вызванного мутацией гена GSN (гельзолин), которое начинается с периферии роговицы в возрасте 30–40 лет, при этом центральная прозрачность сохраняется длительное время. Синдром Меретойя сопровождается такими системными симптомами, как дряблость кожи, маскообразное лицо, периферическая нейропатия и сердечные аритмии2,10). Во втором издании IC3D синдром Меретойя классифицируется отдельно от решетчатой дистрофии роговицы4).
Liu Z, et al. An R124C mutation in TGFBI caused lattice corneal dystrophy type I with a variable phenotype in three Chinese families. Mol Vis. 2008. Figure 2. PMCID: PMC2443752. License: CC BY.
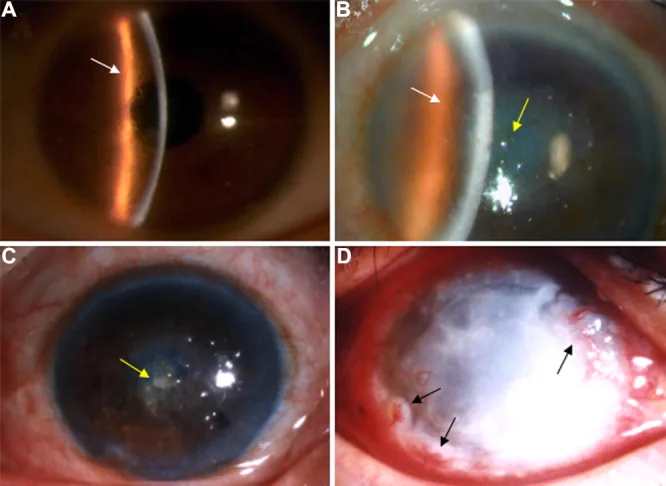
На щелевой лампе видны ветвящиеся решетчатые линии в строме роговицы и помутнение с преобладанием в центре. Это изображение представляет типичные клинические проявления решетчатой дистрофии роговицы.
При LCD1 в детстве большинство пациентов бессимптомны, обнаруживаются лишь мелкие помутнения, выявляемые только при просвечивании щелевой лампой. После 10–20 лет возникают рецидивирующие эрозии роговицы (recurrent corneal erosion, RCE), проявляющиеся резкой болью в глазу, светобоязнью, слезотечением и ощущением инородного тела при пробуждении. К 30 годам в поверхностных слоях стромы центральной части роговицы появляются белые помутнения, а после 40 лет прогрессирует снижение зрения.
При LCD IIIA (вариантный тип) повреждение эпителия обычно не возникает, основной жалобой является медленное снижение зрения после 40 лет.
При старом LCD2 (синдром Меретойя) глазные симптомы появляются в 30–40 лет, но значительное нарушение зрения часто откладывается до 60 лет 11). Часто предшествуют или сопутствуют общие симптомы: дряблость кожи век, маскообразное лицо, прогрессирующая невропатия черепных нервов, сердечные аритмии 2,10).
Ниже приведены данные щелевой лампы для каждого типа заболевания.
LCD1 (классический тип)
Первичная локализация: появляется в зоне зрачка обоих глаз в виде мелких точечных и линейных помутнений в слое Боумена и поверхностной строме.
Решетчатые линии: нитевидные и линейные помутнения с двойным контуром переплетаются, образуя сетчатые и звездчатые помутнения.
Прогрессирующая стадия: в центральной части роговицы возникает желтоватое или круглое молочно-белое помутнение.
Метод ретроградного освещения: полупрозрачные тонкие решетчатые линии, плохо видимые при прямом освещении, становятся четко различимыми.
Окрашивание флуоресцеином: из-за снижения адгезии эпителия поверхность становится шероховатой.
Рецидивирующие эрозии эпителия: возникают часто, так как отложения распространяются на базальные клетки эпителия и мембрану Боумена.
LCD IIIA (вариантный тип)
Решетчатые линии: толстые, длинные решетчатые линии в средних и глубоких слоях стромы, иногда с древовидным ветвлением. Наблюдаются даже при прямом освещении.
Фенотип: существует три паттерна: ① только решетчатые линии, ② только мелкие зернистые отложения, ③ смешанный. В одном и том же организме возможны разные фенотипы на правом и левом глазу, а также односторонние случаи.
Эпителий: обычно поражения эпителия не возникает.
Гомозиготы: у гомозигот L527R решетчатые линии более толстые, а центральные зернистые отложения крупнее, но разница не столь выражена, как между гетеро- и гомозиготами R124H (гранулярный тип II).
GSN-тип (Meretoja)
Решетчатые линии: небольшое количество решетчатых отложений, лишенных тонкости, появляются радиально от периферии.
Центральная прозрачность: центральная область сохраняет прозрачность в течение длительного времени после начала заболевания.
Эрозия эпителия: редка.
Системные проявления: маскообразное лицо, выступающие губы с двигательными нарушениями, отвисшие уши, блефарохалазис и другие изменения внешности 2).
При LCD1 в некоторых случаях центральное круглое помутнение становится особенно выраженным; сообщается о 56-летнем гетерозиготном носителе R124C, которому потребовалась трансплантация роговицы из-за центрального круглого помутнения.
QМожно ли диагностировать LCD1 у ребенка?
A
В детском возрасте LCD1 часто протекает бессимптомно, и аномалии трудно обнаружить при прямом освещении. При детальном осмотре с помощью щелевой лампы в проходящем или отраженном свете можно выявить мелкие точечные или линейные помутнения в поверхностных слоях центральной стромы. У детей с рецидивирующей эрозией роговичного эпителия следует заподозрить LCD1, рекомендуется сбор семейного анамнеза и обследование роговицы родителей. Для подтверждения диагноза полезно генетическое тестирование TGFBI.
Ниже приведены гены и типичные мутации, вызывающие решетчатую дистрофию роговицы.
TGFBI-ассоциированные (LCD1, LCD IIIA, LCD IV)
Локус: 5q31 (ген TGFBI).
Тип наследования: аутосомно-доминантный.
Наиболее частая мутация LCD1: R124C (Arg124Cys) является самой частой5).
Типичные мутации LCD IIIA: описаны L527R (Leu527Arg) и другие. Существуют также гомозиготные случаи.
De novo мутации: сообщалось о случае de novo мутации TGFBI L509P, проявляющейся фенотипом LCD IIIA1). У родителей мутации не было, а у одного из детей мутация унаследовалась1).
Роль TGFBIp: вырабатывается эпителием роговицы, распределяется по всей толщине роговицы, в строме участвует в построении коллагеновых волокон5).
GSN-ассоциированный (синдром Меретойя, ранее LCD2)
Локус: 9q34 (ген GSN, гельзолин).
Тип наследования: аутосомно-доминантный.
Классическая мутация: D187N (финский тип) встречается чаще всего, также описана p.Asp187Tyr10,11).
Новая мутация: p.Glu580Lys, описанная в словенской семье, расположена на границе доменов G4-G5 и вызывает электростатическое отталкивание из-за замены отрицательного заряда на положительный2).
Клиническая картина: помимо решетчатого помутнения роговицы, проявляется системным амилоидозом с дряблостью кожи, сердечными аритмиями, поражением почек и зрительного нерва2).
Поскольку это наследственное заболевание, наиболее важным фактором риска является семейный анамнез. Однако при TGFBI возможны de novo мутации, поэтому отсутствие семейного анамнеза не исключает заболевание1). Тип наследования аутосомно-доминантный: если один из родителей является носителем мутации, вероятность передачи ребенку составляет 50%. Половых различий не наблюдается, расовые различия также не очевидны для LCD1, однако синдром Меретойя известен скоплением семей в Финляндии11).
Вклад факторов окружающей среды неясен; возникновение и прогрессирование заболевания в основном определяется генотипом. Однако частота рецидивирующих эрозий эпителия может увеличиваться в сухой среде, при ношении контактных линз или травмах. Рефракционные операции (LASIK, SMILE и др.) могут вызвать быстрое ухудшение TGFBI-ассоциированных дистрофий, поэтому при предоперационном скрининге следует обращать внимание на пациентов с семейным анамнезом5).
Для дифференциальной диагностики LCD1, вариантных типов и GSN-типа необходимо комплексное использование данных биомикроскопии, гистологии и генетического анализа.
Клинические исследования
Щелевая лампа: при прямом освещении начальные решетчатые линии легко пропустить. При диафаноскопии на фоне зрачка видны мелкие помутнения, а при ретроградном освещении выявляются полупрозрачные тонкие решетчатые линии.
Окрашивание флуоресцеином: при LCD1 из-за снижения адгезии эпителия окрашивание становится грубым. Также полезно для оценки площади эрозии эпителия.
Оптическая когерентная томография переднего сегмента (ОКТ переднего сегмента): позволяет количественно оценить глубину отложений. Измерение глубины поражения с помощью FD-OCT полезно для определения глубины абляции при ПТК1).
Конфокальная микроскопия роговицы: позволяет наблюдать отложения в строме на клеточном уровне.
Окончательный диагноз
Генетическое тестирование: выявление мутаций в генах TGFBI и GSN позволяет установить тип заболевания. Даже при одинаковом фенотипе разные мутации могут влиять на скорость рецидивов и прогрессирования, что напрямую связано с планированием лечения.
Патологическое исследование: окрашивание конго красным дает красный цвет, а под поляризационным микроскопом наблюдается яблочно-зеленое двулучепреломление, что подтверждает амилоид6).
Иммуногистохимия: возможна дифференциация типов с помощью антител к TGFBIp и антител к гельзолину.
Сбор семейного анамнеза: из-за аутосомно-доминантного наследования подтверждение диагноза поддерживается выявлением изменений роговицы у родителей и сибсов.
Гранулярная дистрофия роговицы II типа (тип Авеллино, TGFBI R124H): наиболее частая TGFBI-ассоциированная дистрофия в Японии, проявляется смешанной картиной гранулярных отложений и решетчатых линий. Для дифференциации с LCD1 надежным методом является генетическое тестирование.
Вторичный амилоидоз роговицы: не наследственный, амилоид откладывается вторично на фоне хронического раздражения поверхности глаза, такого как трихиаз или кератоконус. Отличительными признаками являются отсутствие семейного анамнеза и наличие основного заболевания.
Пятнистая дистрофия роговицы: аутосомно-рецессивное наследование, вызванное мутацией гена CHST6, сопровождается диффузным помутнением по типу матового стекла и эндотелиальными аномалиями.
Желатинозная каплевидная дистрофия роговицы: аутосомно-рецессивное наследование, вызванное мутацией гена TACSTD2, проявляется молочно-белыми желатинозными возвышениями. Относительно часто встречается в Японии.
QПочему генетическое тестирование важно?
A
Даже если фенотип решетчатой дистрофии роговицы схож, различия в гене-возбудителе и месте мутации приводят к значительным изменениям в скорости прогрессирования, частоте рецидивов, выборе лечения и наличии системных осложнений. Лечебная тактика и необходимость системного обследования принципиально различаются между LCD1 с мутацией TGFBI и синдромом Меретойя с мутацией GSN2,10). Кроме того, описаны случаи de novo мутаций, когда только семейный анамнез не позволяет определить тип заболевания1), поэтому генетическое тестирование необходимо для окончательного диагноза и классификации.
В детском и молодом возрасте при отсутствии симптомов или наличии только мелких помутнений рекомендуется наблюдение. Оценка прогрессирования проводится с помощью щелевой лампы каждые 6–12 месяцев.
При рецидивирующей эрозии роговицы, являющейся основным симптомом LCD1, первой линией является следующая консервативная терапия:
Лечение приступов: непрерывное ношение лечебных мягких контактных линз для защиты эпителия роговицы. Одновременно применяются антибактериальные капли для профилактики вторичной инфекции. Нанесение глазной мази для увлажнения и защиты эпителия.
Профилактика рецидивов: применение глазной мази перед сном подавляет рецидивы приступов RCE. В сухой среде также рекомендуется использовать искусственные слезы или смазывающие средства в дневное время.
При LCD1, где преобладают поверхностные отложения амилоида, в случае выраженного центрального помутнения или повторяющихся рецидивов эрозии роговицы, методом первого выбора является фототерапевтическая кератэктомия (PTK) с использованием эксимерного лазера7,8). Обычно ранних рецидивов не возникает, но со временем рецидив неизбежен, и PTK может быть выполнена на одном глазу до двух раз.
У гетерозигот рецидив протекает медленно, и повторное лечение требуется редко. У гомозигот рецидив имеет тенденцию возникать раньше, чем у гетерозигот. Частота рецидивов после PTK, как и при других дистрофиях, связанных с TGFBI, увеличивается со временем, и при длительном наблюдении у многих пациентов выявляются некоторые признаки рецидива8).
В качестве примера эффективности PTK: у пациента с LCD IIIA, вызванной de novo мутацией TGFBI L509P, была выполнена PTK на глубину 60 мкм под контролем FD-OCT, и максимально корригированная острота зрения (BCVA) улучшилась с 20/400 до 20/501). Через 45 месяцев после операции не было отмечено снижения зрения или значимого рецидива1).
Согласно Preferred Practice Pattern по отеку и помутнению роговицы AAO, PTK при гранулярной и решетчатой дистрофиях роговицы является «разумным вариантом» и может отсрочить переход к DALK или сквозной кератопластике, но существует риск послеоперационного хейза. При повторных процедурах рассматривается применение митомицина C для подавления рецидивирующих рубцов и стромальных отложений. Предупреждается, что при абляции, превышающей переднюю треть стромы, или при остаточной толщине ложа менее 250 мкм, риск кератэктазии возрастает7).
При рецидивирующих случаях или когда помутнение распространяется глубже средней стромы, выбирают трансплантацию роговицы. При LCD1 обычно до 40 лет трансплантация роговицы не требуется. Поскольку эндотелий роговицы при LCD в норме, метод выбирают в зависимости от глубины помутнения.
Высокое восстановление зрения, но риск отторжения и рецидива
В последние годы DALK широко используется как новый метод первой линии благодаря снижению риска отторжения и результатам остроты зрения, сопоставимым с полнослойной кератопластикой.
Рецидив LCD после пересадки роговицы неизбежен; сообщается, что частота рецидивов после полнослойной кератопластики составляет 17,8% через 5 лет, 26% через 8 лет и 56% через 15 лет9). Поскольку рецидивное помутнение обычно ограничивается поверхностными слоями, его можно удалить с помощью PTK, что продлевает время до повторной трансплантации. При LCD IIIA (вариантный тип) лечение часто не требуется, если только не происходит значительного влияния на зрение.
QНасколько эффективна PTK?
A
PTK позволяет эффективно удалять поверхностные отложения амилоида, улучшая зрение и уменьшая рецидивирующие эрозии эпителия. Сообщается, что у пациента с LCD IIIA после PTK на глубину 60 мкм максимально корригированная острота зрения улучшилась с 20/400 до 20/50, и рецидива не было в течение 45 месяцев1). У гетерозигот рецидив медленный, но у гомозигот наблюдается ранний рецидив. Глубокие поражения не могут быть удалены с помощью PTK, поэтому при глубоких помутнениях требуется DALK или полнослойная кератопластика7).
QВозникает ли рецидив после пересадки роговицы?
A
Рецидив LCD после пересадки роговицы неизбежен. Сообщается, что частота рецидивов после полнослойной кератопластики составляет 17,8% через 5 лет, 26% через 8 лет и 56% через 15 лет9). Однако рецидивное помутнение обычно ограничивается поверхностными слоями трансплантата, поэтому его можно удалить с помощью PTK, продлевая срок службы трансплантата. Глубокая послойная кератопластика (DALK) имеет более низкий риск эндотелиального отторжения по сравнению с полнослойной кератопластикой и привлекает внимание как новый метод первой линии7).
Центральным звеном патогенеза LCD1 является аномальное накопление TGFBIp (kerato-epithelin, βig-h3). TGFBIp в норме продуцируется эпителием роговицы, распределяется по всей ее толщине и в строме участвует в построении коллагеновых волокон и клеточной адгезии5). Аномальный белок, образующийся в результате мутации R124C, подвергается неправильному сворачиванию и самоагрегации, откладываясь в виде нерастворимых амилоидных фибрилл в слое Боумена и поверхностных слоях стромы. На поздних стадиях отложения распространяются в глубокие слои стромы.
Амилоидные отложения вызывают изменения в структуре адгезии эпителия передней роговицы, приводя к дегенерации базальных клеток эпителия и дегенерации эпителиального слоя с дефектами мембраны Боумена. Этот структурный разрыв является патогенетической основой рецидивирующей эрозии роговицы.
Фенотипическая дифференциация в зависимости от генотипа TGFBI
В гене TGFBI различия в сайте мутации и замещаемой аминокислоте определяют клиническую картину. R124C вызывает LCD1, R124H — гранулярную дистрофию роговицы II типа (тип Авеллино), R124L — дистрофию роговицы Рейс-Бюклерса5). Молекулярный механизм, при котором различие всего в одной аминокислоте определяет характер отложений (амилоид vs гиалин vs оба) и их локализацию, полностью не выяснен, но считается, что ключевую роль играет принадлежность мутированного участка к определенному домену βig-h3 и влияние на стабильность укладки.
При LCD IIIA мутации, такие как L527R, с преимущественным поражением глубоких слоев вызывают толстые веревкообразные решетчатые линии и позднюю форму без поражения эпителия. Послойная локализация отложений может объясняться градиентом секреции и диффузии βig-h3 от продуцирующих клеток (базальных эпителиоцитов) в строму и различиями в стабильности укладки мутантного белка. Считается, что R124C предпочитает путь от промежуточных форм укладки к образованию амилоидных фибрилл, накапливая амилоид вокруг слоя Боумена5). Напротив, мутация L527R образует относительно стабильный неправильно свернутый белок, который медленно откладывается в более глубоких слоях стромы.
Задний роговичный амилоид и риск внутриглазных операций
Ранее считалось, что амилоидные отложения при LCD1 ограничены передней роговицей (слой Боумена — поверхностные слои стромы). Однако недавние патологические исследования показали наличие амилоидных отложений также в задней роговице вблизи десцеметовой мембраны3). Амилоидные отложения в задней роговице могут влиять на адгезию десцеметовой мембраны и способствовать ее отслойке во время операции по удалению катаракты3). Предполагается, что в задней роговице действует тот же механизм, что и в передней, где амилоидные отложения нарушают адгезию эпителия3).
Гельсолин, молекула, вызывающая старый LCD2 (синдром Меретойя), присутствует как в цитоплазме, так и вне клеток, и является белком, участвующим в клеточной подвижности, делении и апоптозе через связывание актина. Классическая мутация D187N, называемая финским типом, проявляется решетчатыми отложениями в роговице и черепно-мозговыми нарушениями11). Новая мутация p.Glu580Lys, описанная в словенской семье, расположена на границе доменов G4-G5 и вызывает электростатическое отталкивание из-за замены отрицательно заряженного глутамата на положительно заряженный лизин, что снижает междоменную связь и стабильность2). Мутантный гельсолин подвергается аномальному расщеплению фурином и MT1-MMP в плазме, высвобождая амилоидные предшественники размером 8 кДа и 5 кДа. Они откладываются в строме роговицы, коже, сосудистой стенке, периферических нервах и почечных клубочках, вызывая мультиорганные симптомы, характерные для синдрома Меретойя2,11). Отложения в роговице часто предшествуют другим системным проявлениям, что может позволить офтальмологу первым диагностировать это заболевание.
Сообщается о возникновении LCD вследствие de novo мутаций в гене TGFBI1). Даже при отсутствии семейного анамнеза следует учитывать возможность de novo мутации, и рекомендуется подтверждение с помощью генетического тестирования1). Мутация L509P редка, но проявляется разнообразными фенотипами от дистрофии роговицы Рейс-Бюклерса до LCD IIIA1).
В гене GSN, помимо традиционной мутации p.Asp187Asn/Tyr, описана новая мутация p.Glu580Lys, которая вызывает системный амилоидоз с решетчатой дистрофией роговицы, дряблостью кожи, сердечными аритмиями, поражением почек и оптической нейропатией2).
Задние поражения роговицы и ведение интраокулярной хирургии
Патологически показано, что у пациентов с LCD1 в задней части роговицы присутствуют амилоидные отложения, которые могут влиять на адгезию десцеметовой мембраны3). Необходимо обращать внимание на риск отслойки десцеметовой мембраны при интраокулярных операциях, включая хирургию катаракты.
Это открытие имеет клиническое значение для оценки показаний к хирургии катаракты и планирования оперативной техники у пациентов с LCD1.
Разрабатываются более точные хирургические методы, такие как фемтосекундная лазерная послойная кератэктомия (FLK) и фемтосекундная лазерная послойная кератопластика (FALK)12). Они позиционируются как дополнение к традиционной PTK благодаря улучшенной гладкости поверхности среза и воспроизводимому контролю глубины.
Поскольку мутации TGFBI являются аутосомно-доминантными с усилением функции, на доклинической стадии изучаются аллель-специфичные siRNA, антисмысловые олигонуклеотиды и аллель-специфичный нокаут с помощью CRISPR-Cas9. Роговица является благоприятной мишенью для генной терапии благодаря возможности местного введения и иммунной привилегии. Однако в настоящее время ни один из методов не применяется клинически, и все они требуют дальнейшей проверки долгосрочной безопасности и эффективности.
Низкомолекулярные соединения, нацеленные на агрегацию TGFBIp и мутантного гельзолина, молекулярные шапероны (например, индукторы Hsp70) и ингибиторы связывания амилоидных фибрилл изучаются на фундаментальном уровне. Для системного гельзолинового амилоидоза некоторые препараты, подавляющие расщепление мутантного гельзолина в плазме, оцениваются в доклинических испытаниях2). В будущем такая таргетная терапия может стать радикальным лечением, заменяющим традиционное физическое удаление (PTK и трансплантацию роговицы).
Протеомный анализ роговицы с помощью масс-спектрометрии показал, что в отложениях при LCD1 могут сосуществовать не только TGFBIp, но и несколько аномальных белков. Для будущего клинического применения изучается вклад этих совместно осаждаемых белков в патогенез.
Ji YW, Ahn H, Shin KJ, Kim TI, Seo KY, Stulting RD, et al. De Novo L509P Mutation of the TGFBI Gene Associated with Slit-Lamp Findings of Lattice Corneal Dystrophy Type IIIA. Journal of clinical medicine. 2022;11(11). doi:10.3390/jcm11113055. PMID:35683443; PMCID:PMC9181583.
Potrc M, Volk M, de Rosa M, Pizem J, Teran N, Jaklic H, Maver A, Drnovsek-Olup B, Bollati M, Vogelnik K, Hocevar A, Gornik A, Pfeifer V, Peterlin B, Hawlina M, Fakin A. Clinical and Histopathological Features of Gelsolin Amyloidosis Associated with a Novel GSN Variant p.Glu580Lys. Int J Mol Sci. 2021;22(3):1084.
Matsumoto A, Fukuoka H, Sotozono C. Descemet’s Membrane Detachment During Cataract Surgery in Lattice Corneal Dystrophy Type I: Histopathological Analysis of Posterior Corneal Involvement. Cureus. 2025;17(3):e81431. doi:10.7759/cureus.81431. PMID:40296953; PMCID:PMC12037203.
Weiss JS, Møller HU, Aldave AJ, et al. IC3D classification of corneal dystrophies-Edition 2. Cornea. 2015;34(2):117-159. doi:10.1097/ICO.0000000000000307. PMID:25564336.
Han KE, Choi SI, Kim TI, Maeng YS, Stulting RD, Ji YW, et al. Pathogenesis and treatments of TGFBI corneal dystrophies. Progress in retinal and eye research. 2016;50:67-88. doi:10.1016/j.preteyeres.2015.11.002. PMID:26612778.
Lisch W, Seitz B. The clinical landmarks of corneal dystrophies. Developments in ophthalmology. 2011;48:9-23. doi:10.1159/000324075. PMID:21540629.
American Academy of Ophthalmology Cornea/External Disease Panel. Corneal Edema and Opacification Preferred Practice Pattern. San Francisco, CA: American Academy of Ophthalmology; 2024.
Dinh R, Rapuano CJ, Cohen EJ, Laibson PR. Recurrence of corneal dystrophy after excimer laser phototherapeutic keratectomy. Ophthalmology. 1999;106(8):1490-1497.
Marcon AS, Cohen EJ, Rapuano CJ, Laibson PR. Recurrence of corneal stromal dystrophies after penetrating keratoplasty. Cornea. 2003;22(1):19-21.
Meretoja J. Familial systemic paramyloidosis with lattice dystrophy of the cornea, progressive cranial neuropathy, skin changes and various internal symptoms. A previously unrecognized heritable syndrome. Ann Clin Res. 1969;1(4):314-324. PMID:4313418.
Kivelä T, Tarkkanen A, Frangione B, Ghiso J, Haltia M. Ocular amyloid deposition in familial amyloidosis, Finnish: an analysis of native and variant gelsolin in Meretoja’s syndrome. Invest Ophthalmol Vis Sci. 1994;35(10):3759-3769. PMID:8088963.
Steger B, Romano V, Biddolph S, Willoughby CE, Batterbury M, Kaye SB. Femtosecond laser-assisted lamellar keratectomy for corneal opacities secondary to anterior corneal dystrophies: an interventional case series. Cornea. 2016;35(1):6-13. PMID:26509759. doi:10.1097/ICO.0000000000000665.
Скопируйте текст статьи и вставьте его в выбранный ИИ-ассистент.
Статья скопирована в буфер обмена
Откройте ИИ-ассистент ниже и вставьте скопированный текст в чат.