Die Fuchs-Endotheldystrophie (FECD) ist eine fortschreitende Erkrankung, bei der die Hornhautendothelzellen beidseitig abnormal werden. 1910 berichtete Ernst Fuchs erstmals über 13 Fälle als „dystrophia epithelialis corneae“, und später wurde festgestellt, dass es sich um eine Endothelperkrankung handelt, woraus der heutige Name entstand 1).
Auf der zentralen Endotheloberfläche der Hornhaut treten guttae (guttata) auf, die sich allmählich zur Peripherie ausbreiten. Wenn die Barriere- und Pumpfunktion (Na⁺/K⁺-ATPase) der Endothelzellen nachlässt, kommt es zu einem Ödem des Hornhautstromas, gefolgt von Epithelödem und Blasenbildung. Die Descemet-Membran verdickt sich und wird unregelmäßig, was zur Trübung der Hornhaut führt.
In der 2. Auflage der IC3D (Internationale Klassifikation der Hornhautdystrophien) (Weiss 2015) wird die FECD in die Kategorie „Hornhaut-Endotheldystrophien“ eingeordnet 15). Je nach Erkrankungsalter werden zwei Haupttypen unterschieden.
Frühmanifestation (FECD1): Tritt im Kindes- oder Jugendalter auf. Hauptursache sind Punktmutationen (L450W, Q455K usw.) im COL8A2-Gen (1p34.3-p32.3), das die α2-Kette von Kollagen Typ VIII kodiert 1).
Spätmanifestation (FECD2 und folgende): Tritt langsam im 5.–6. Lebensjahrzehnt auf. Die häufigste Ursache ist eine CTG-Trinukleotid-Repeat-Expansion im TCF4-Gen (79 % in westlichen Ländern) 1).
Epidemiologie (Japan im internationalen Vergleich)
Häufigkeit von Cornea guttata bei Patienten vor Kataraktoperation
1,2 %
Nationale multizentrische Erhebung
Prävalenz in Japan (Kumejima-Studie) bei ≥40-Jährigen
4,1 %
Higa 20117)
Prävalenz bei japanischen Frauen (≥40 Jahre)
5,8 %
Higa 20117)
Prävalenz bei Männern in Japan (40 Jahre und älter)
2,4 %
Higa 20117)
Island - Reykjavik Eye Study, 55 Jahre und älter
Frauen 11 %, Männer 7 %
Zoega 200610)
Geschlechterverhältnis (international)
2,5:1 bis 3,5:1 (weiblich dominant)
Matthaei 20191)
Häufigkeit der TCF4-Repeat-Expansion bei Japanern
12 von 47 Fällen (26 %)
Nakano 20158)
Japaner als gelbe Rasse haben tendenziell eine geringere Inzidenz von FECD als Kaukasier und Schwarze. Mit der alternden Gesellschaft in Japan wird jedoch ein weiterer Anstieg erwartet. Es wird angenommen, dass Japaner eine höhere Dichte an Hornhautendothelzellen haben als Kaukasier, was den Ausbruch der Erkrankung relativ verzögert.
Bei vielen Japanern mit engen Kammerwinkeln ist eine Abnahme der Endothelzellen nach einer Laser-Iridotomie (LI) nicht selten, und eine frühzeitige Erkennung der FECD erfordert besondere Aufmerksamkeit.
QWie häufig tritt diese Erkrankung auf?
A
In einer Bevölkerungsstudie auf Okinawa, Insel Kumejima (Kumejima Study), wurden bei 4,1 % der Personen ab 40 Jahren Hornhaut-Guttae festgestellt. Bei Frauen waren es 5,8 %, bei Männern 2,4 % 7). Es gibt auch nationale Daten, dass 1,2 % der Patienten, die vor einer Kataraktoperation untersucht wurden, eine Guttata-Hornhaut aufweisen. Die Häufigkeit wird bei Japanern als geringer angesehen als bei Europäern oder Amerikanern, nimmt aber mit der alternden Gesellschaft zu.
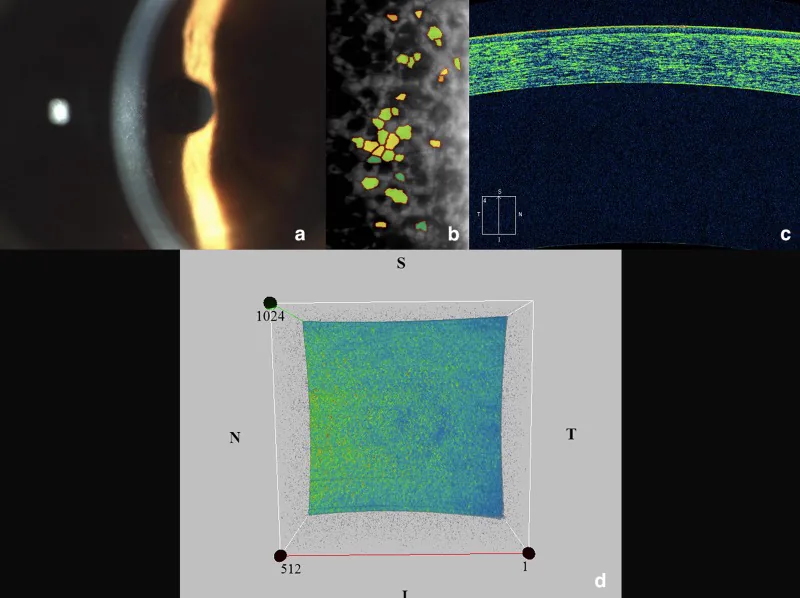
Iovino C, et al. Corneal endothelium features in Fuchs’ Endothelial Corneal Dystrophy: A preliminary 3D anterior segment optical coherence tomography study. PLoS One. 2018. Figure 2. PMCID: PMC6264151. License: CC BY.
Ein zusammengesetztes Bild, das die Hornhautendothel-Anomalien der Fuchs-Endotheldystrophie zeigt. Die Spaltlampe zeigt ein gehämmertes Metall-Aussehen, Spekular- und AS-OCT zeigen Endothelzellverlust und hyperreflektive Punkte auf der hinteren Hornhautoberfläche.
In der Regel verläuft die Erkrankung vor dem 50. Lebensjahr asymptomatisch. Die Symptome schreiten langsam fort, korrelierend mit dem Grad des Ödems.
Morgendliches Verschwommensehen: Das Hornhautödem verschlimmert sich durch die nächtliche Lidspalte, was zu einer stärksten Sehverschlechterung direkt nach dem Aufwachen führt. Tagsüber bessert sich das Ödem durch geöffnete Lider, und am Abend erholt sich das Sehen, ein charakteristisches Muster 1).
Anhaltende Sehverschlechterung: Bei schwerem Ödem bleibt die Sehverschlechterung den ganzen Tag über bestehen 2).
Photophobie und Blendung: Verstärkt durch Lichtstreuung an der unregelmäßigen Descemet-Membran1).
Augenschmerzen und Tränenfluss: Bei schwerem Epithelödem bilden sich Blasen, deren Platzen starke Augenschmerzen und Tränenfluss verursacht 1).
Klinische Befunde (vom Arzt bei der Untersuchung festgestellt)
Symptome: Schwere Sehverschlechterung den ganzen Tag, Augenschmerzen, Tränenfluss. Deutlich eingeschränkte Lebensqualität.
Diese klinische Stadieneinteilung basiert auf der modifizierten Klassifikation nach Krachmer et al. (1978)5).
Details der Spaltlampenbefunde:
Guttae: Halbkugelförmige Vorwölbungen aus abnormalem kollagenähnlichem Material, das von degenerierten Endothelzellen produziert wird, auf der Rückseite der Descemet-Membran in die Vorderkammer. Als grau-weiße oder bräunliche Körnchen auf der Hornhautrückfläche sichtbar.
„Beaten-Metal“-Aussehen: Charakteristisches Erscheinungsbild durch Konfluenz und Zunahme der Guttae kombiniert mit Pigmentierung. Am besten im Spiegelbezirk zu beobachten.
Hornhautödem: Fortschreiten von Unregelmäßigkeit der Descemet-Membran → Stromaquellung (Stromaödem) → subepitheliale Flüssigkeitsansammlung (Epithelödem).
QWarum sieht man morgens schlecht und tagsüber besser?
A
In einer gesunden Hornhaut pumpen die Endothelzellen ständig Wasser in die Vorderkammer, um die Transparenz zu erhalten. Bei FECD ist die Pumpfunktion des Endothels vermindert, sodass während des Schlafens (geschlossene Augenlider) auch die Wasserverdunstung aufhört und die Hornhaut morgens am stärksten ödematös ist, mit verstärktem Nebelsehen. Bei geöffneten Augen verdunstet Wasser von der Hornhautoberfläche, und tagsüber bessert sich das Ödem etwas, sodass die Sehkraft zurückkehrt. Mit fortschreitender Erkrankung verschwindet diese tageszeitliche Schwankung und das Nebelsehen hält den ganzen Tag an.
FECD wird hauptsächlich autosomal-dominant vererbt, aber es gibt Variationen in Penetranz und Expressivität, und einige Fälle haben keine eindeutige Familienanamnese.
Hauptverursachende Gene :
TCF4-Gen (18q21.2) : Die Expansion der CTG-Trinukleotid-Wiederholung (CTG18.1) ist die häufigste Ursache. Mehr als 50 Wiederholungen gelten als pathologisch und werden bei etwa 79 % der westlichen FECD-Patienten nachgewiesen1,9). Bei 47 japanischen FECD-Patienten wurde sie bei 12 (26 %) gefunden, eine niedrigere Häufigkeit als im Westen8); im Westen korreliert sie mit dem Schweregrad, bei Japanern jedoch kaum1).
COL8A2-Gen (1p34.3-p32.3) : Kodiert für die α2-Kette von Kollagen Typ VIII. Punktmutationen wie L450W, Q455K, Q455V verursachen die früh beginnende Form (FECD1)1).
Weitere Kandidatengene : SLC4A11, TCF8/ZEB1, AGBL1, LOXHD1, TGFBI, CLU wurden berichtet1).
Bei Japanern ist die Häufigkeit der TCF4-Wiederholungsexpansion niedriger als im Westen, daher ist die Aufklärung anderer genetischer Hintergründe erforderlich8).
Systemische Begleiterkrankungen: Assoziation mit myotoner Dystrophie Typ 1 (DM1) wurde berichtet1)
QIst es erblich? Betrifft es Kinder?
A
FECD wird hauptsächlich autosomal-dominant vererbt. Theoretisch beträgt die Wahrscheinlichkeit einer Vererbung an ein Kind 50 %. Allerdings variieren Beginn und Schweregrad stark (unvollständige Penetranz); viele Menschen, die das Gen erben, haben lebenslang nur sehr milde Symptome. Insbesondere bei Japanern ist der Anteil der TCF4-Genmutation, der häufigsten Ursache bei Westlern, gering8), was auf einen anderen genetischen Hintergrund hindeutet. Bei Bedenken wird eine Konsultation mit einem Genetiker empfohlen.
Beurteilung von Guttae und Ödem der hinteren Hornhautfläche bei direkter Beleuchtung.
Die Spiegelreflexmethode ist am wichtigsten, unverzichtbar zur Bestätigung des „beaten-metal“-Aspekts.
Beurteilung des Ausmaßes von Stromaödem und -trübung bei retrograder Beleuchtung.
Beachten Sie, dass die Endothelzelldichte der Hornhaut bei Japanern höher ist als bei Weißen, sodass Symptome bei gleichem Endothelverlust weniger ausgeprägt sein können.
Dies ist die wichtigste Untersuchung für die Diagnose und Nachsorge der FECD.
Parameter
Normalwert
Grenzwert für Anomalie
Endothelzellendichte (Neugeborenenperiode)
3.500–4.000 Zellen/mm²
—
Endothelzellendichte (20 Jahre)
2.700 Zellen/mm²
—
Endothelzellendichte (70 Jahre und älter)
Durchschnitt 2.200 Zellen/mm²
—
Grenze der Transparenzerhaltung
—
400–500 Zellen/mm² oder weniger
CV-Wert (Variationskoeffizient)
0,2–0,3
≥ 0,35
Hexagonalitätsrate (hexagonality)
60–70 %
≤ 50 %
dunkler Fleck: Die Erhebungen der Guttae weichen von der Ebene des Spiegelreflexes ab und werden auf dem Spekulum als schwarze kreisförmige Bereiche beobachtet. Die Endothelzellen fehlen nicht tatsächlich, sondern sind aufgrund der Erhebungen nicht in derselben Ebene sichtbar.
Bei schwerem Ödem oder Trübung ist das Kontakt-Spekularmikroskop nützlicher als das berührungslose, da es umfangreichere und klarere Endothelbilder liefert.
Die normale Endothelzellverlustrate beträgt 0,5 %/Jahr. Nach Kataraktoperation sind es 2 %/Jahr, nach Glaukomoperation beschleunigt sie sich auf 10 %/Jahr.
Konfokale Mikroskopie: Ermöglicht die schichtweise Beobachtung aller Hornhautschichten. Die Morphologie der Guttae und Details der Descemet-Membran können beurteilt werden1).
Vorderabschnitts-OCT: Ermöglicht die nicht-invasive Quantifizierung der Hornhautdicke, der Verdickung der Descemet-Membran und des subepithelialen Ödems1).
Ultraschall-Pachymetrie (Hornhautdickenmessung): Goldstandard für die präoperative Beurteilung. Eine zentrale Hornhautdicke >640 μm ist ein Hinweis auf ein erhöhtes Risiko einer postoperativen Hornhautdekompensation1).
Scheimpflug-Bildgebung: Ermöglicht die Beurteilung des Dickenverhältnisses von Zentrum zu Peripherie (central-to-peripheral thickness ratio)1).
Modifizierte Krachmer-Klassifikation (Krachmer et al. 1978)5): Wird zur Bestimmung des Krankheitsstadiums und der Operationsindikation verwendet.
PEX-Materialablagerungen, erhöhter Augeninnendruck, PEX-Material auf der Linsenvorderseite als Schlüssel zur Differenzialdiagnose
ICE (iridokorneales endotheliales) Syndrom
Einseitig, mit Irisatrophie, vorderen Synechien und Glaukom. Keine Guttae
Endothelveränderungen bei Engwinkelaugen
Kann guttaatähnliche Befunde zeigen. Differenzierung durch Augeninnendruck und Kammerwinkelmorphologie
QWas ist ein Spekularmikroskop? Was kann man damit feststellen?
A
Das Spekularmikroskop (Spiegelreflex-Endothelzell-Fotografiegerät) ist ein Gerät, das die Endothelzellen der innersten Schicht der Hornhaut mittels spezieller Lichtreflexion nichtinvasiv fotografiert und vermisst. Die Untersuchung misst die Anzahl der Endothelzellen (Zelldichte), die Größenvarianz (CV-Wert) und die Formgleichmäßigkeit (Prozentsatz sechseckiger Zellen). Bei der FECD erscheinen die Guttae als schwarze Punkte (dunkle Flecken), was zur Beurteilung des Krankheitsstadiums beiträgt. Die Aufnahme dauert einige Minuten und ist schmerzfrei.
Das Ziel der Behandlung ist die Wiederherstellung der Hornhauttransparenz und die Erhaltung des Sehvermögens. Je nach Krankheitsstadium wird eine symptomatische oder operative Therapie gewählt.
Sie dient der Linderung der Symptome vor einer Operationsindikation. Sie hat keine Wirkung auf die Wiederherstellung der Endothelzellzahl oder die Verlangsamung des Krankheitsverlaufs.
5% hypertonische Kochsalzlösung Augentropfen/Salbe: Nutzt den osmotischen Druckunterschied, um Wasser aus der Hornhaut zu ziehen und Ödeme zu reduzieren. Hauptsächlich nützlich zur Linderung von morgendlichem verschwommenem Sehen.
Therapeutische Kontaktlinsen: Werden getragen, um Augenschmerzen und Tränenfluss durch das Platzen von Blasen zu reduzieren.
Hornhauttrocknung mit einem Haartrockner: Warme Luft wird auf das geschlossene Auge gerichtet, um die Verdunstung von Wasser von der Hornhautoberfläche zu fördern 1). Führt zu einer vorübergehenden Besserung des Ödems.
In einer Metaanalyse von Sela et al. (2023) mit 8 Studien (376 Augen) war der BCVA nach 12 Monaten bei DMEK signifikant besser (−0,06 logMAR)3). Auch die multizentrische RCT von Dunker et al. (2020) zeigte eine höhere Rate von ≥20/25 bei DMEK im Vergleich zu UT-DSAEK (66% vs. 33%, p=0,02)4). Bei UT-DSAEK-Transplantaten mit einer Dicke unter 70 μm verringerte sich der Unterschied zur DMEK jedoch3).
Die Gruppe der Universität Kyoto (Kinoshita 2018) entwickelte eine Therapie, bei der kultivierte gesunde Spender-Hornhautendothelzellen zusammen mit einem ROCK-Inhibitor (Y-27632) in die Vorderkammer injiziert werden13).
24 Wochen postoperativ erholte sich die Zelldichte bei 10 von 11 Augen (91%) auf > 1.000 Zellen/mm²
Bei 10 von 11 Augen verbesserte sich die Hornhautdicke auf <630 μm
Kein Transplantat erforderlich; Möglichkeit, viele Patienten mit wenigen Spenderzellen zu behandeln
Der ROCK-Inhibitor wirkt, indem er die Adhäsion von Endothelzellen fördert, Apoptose hemmt und den Zellzyklus vorantreibt13).
Bei FECD tritt häufig eine Katarakt auf, daher ist bei der Wahl des Operationszeitpunkts und -verfahrens Vorsicht geboten.
Eine präoperative zentrale Hornhautdicke >640 μm ist mit einem hohen Risiko für eine Hornhautdekompensation nach alleiniger Kataraktoperation verbunden; daher wird eine gleichzeitige Operation mit Endotheltransplantation empfohlen1,16).
Bei Krachmer-Grad 2,5–4 benötigen etwa 20% der Patienten nach alleiniger Kataraktoperation eine Endotheltransplantation; eine gleichzeitige Operation wird empfohlen1).
Während der Operation werden Endothelschutztechniken mit viskoelastischen Substanzen wie der Soft-Shell-Methode angewendet1).
QSollte man DMEK oder DSAEK wählen?
A
DMEK bietet aufgrund des sehr dünnen Transplantats (ca. 15 μm) eine schnellere Visuserholung und weniger postoperative Refraktionsänderungen. Metaanalysen zeigen auch einen besseren BCVA nach 12 Monaten für DMEK3). DSAEK hingegen ist etwas einfacher zu handhaben, hat eine kürzere Lernkurve für den Chirurgen und wird in Japan häufig durchgeführt. Es gibt Berichte, dass ultradünne DSAEK (<70 μm) nahezu gleichwertige Visusergebnisse wie DMEK erzielt3). Die Wahl hängt von der Erfahrung des Chirurgen, der Erfahrung der Einrichtung und dem Zustand der Hornhaut des Patienten ab. Beide Verfahren sind in Japan seit 2016 (DMEK) bzw. 2009 (DSAEK) krankenversichert.
6. Pathophysiologie und detaillierte Krankheitsmechanismen
Normale Hornhautendothelzellen teilen sich in der Vorderkammer nicht. Bei einem Endotheldefekt vergrößern und wandern benachbarte Zellen, um den Defekt zu decken, sodass die Zelldichte mit dem Alter irreversibel abnimmt. Unter 400–500 Zellen/mm² wird die Aufrechterhaltung der Hornhauttransparenz schwierig.
Bei FECD produzieren und lagern degenerierte Endothelzellen abnormale kollagenähnliche Substanzen auf der Rückseite der Descemet-Membran ab und bilden Guttae. Die Descemet-Membran verdickt und wird unregelmäßig, was einen Teufelskreis schafft, der die Endothelfunktion weiter beeinträchtigt.
Oxidativer Stressweg: Produktion reaktiver Sauerstoffspezies (ROS) durch UV, Rauchen und Alterung → mitochondriale Dysfunktion → weitere ROS-Produktion → DNA-Schäden und Apoptose
Endoplasmatischer Retikulum (ER)-Stressweg: Akkumulation mutierter Proteine (wie COL8A2) im ER → Aktivierung der ungefalteten Proteinantwort (UPR) → Förderung der Apoptose
Endothel-Mesenchym-Transition (EndMT): Umwandlung von Endothelzellen in fibroblastenähnliche Zellen → abnormale Ablagerung extrazellulärer Matrix (ECM) → Förderung der Guttae-Bildung
Sekundärer Stress durch Guttae: Mechanische Schädigung und Kontaktstress durch Guttae → weitere Apoptose verbleibender Endothelzellen → Beschleunigung des Teufelskreises
Alterung, UV-Exposition und Rauchen erhöhen alle den oxidativen Stress und dienen als Eintrittspforte in den Teufelskreis2).
Molekularer Mechanismus der TCF4-CTG-Repeat-Expansion1)
Die Pumpfunktion des Hornhautendothels ist von der Na⁺/K⁺-ATPase abhängig. Wenn die Endothelzellen geschädigt sind, kommt es auf folgenden Wegen zu einem Ödem.
Verminderte endotheliale Pumpfunktion → Wasserbewegung vom Kammerwasser in das Hornhautstroma → Stromaquellung (Stromaödem)
Schweres Stromaödem → Flüssigkeitsansammlung unter dem Epithel → Epithelödem → Blasenbildung → Schmerzen durch Ruptur
Bei einem Anstieg des Augeninnendrucks (Augenhochdruck), der den Quellungsdruck des Hornhautstromas übersteigt, kann es auch bei relativ gesundem Endothel zu einem Epithelödem kommen, was beachtet werden muss.
Antisense-Oligonukleotid (ASO)-Therapie: Zielgerichtet auf TCF4-CTG-Wiederholungs-abgeleitete RNA-Foci, mit dem Ziel der Beseitigung nukleärer Foci, Freisetzung von MBNL1 und Normalisierung des fehlerhaften Spleißens (Hu 2018, Zarouchlioti 2018)1).
Therapie zur Reduktion von oxidativem Stress: Antioxidantien wie NAC (N-Acetylcystein), Lithium und Sulforaphan werden als Kandidaten untersucht1).
Verbreitung der regenerativen Medizin und Zelltherapie
Multizentrische Ausweitung der Therapie mit injizierten kultivierten humanen Hornhautendothelzellen (Kyoto-Protokoll)13). Es könnte möglich sein, eine große Anzahl von Patienten mit wenigen Spenderhornhäuten zu behandeln, und es wird als Lösung für den Spendermangel erwartet.
Erweiterung der Indikationen von ROCK-Inhibitoren (Ripasudil, Y-27632) allein oder als adjuvante Therapie nach DSO14).
Forschung zur Entwicklung eines Frühdiagnose-Scoring-Systems, das Genotyp (TCF4-Wiederholungsanzahl), Geschlecht, Alter, ethnische Zugehörigkeit und Rauchergeschichte kombiniert1).
Aufklärung der Pathologie und Wirkstoffscreening mittels UV-induziertem In-vivo-Mausmodell2).
Da der Beitrag der TCF4-Repeat-Expansion bei Japanern relativ gering ist8), ist die Aufklärung der für Japaner spezifischen genetischen und umweltbedingten Hintergründe eine wichtige zukünftige Aufgabe.
Matthaei M, Hribek A, Clahsen T, Bachmann B, Cursiefen C, Jun AS. Fuchs Endothelial Corneal Dystrophy: Clinical, Genetic, Pathophysiologic, and Therapeutic Aspects. Annual review of vision science. 2019;5:151-175. doi:10.1146/annurev-vision-091718-014852. PMID:31525145.
Ong Tone S, Kocaba V, Böhm M, Wylegala A, White TL, Jurkunas UV. Fuchs endothelial corneal dystrophy: The vicious cycle of Fuchs pathogenesis. Progress in retinal and eye research. 2021;80:100863. doi:10.1016/j.preteyeres.2020.100863. PMID:32438095; PMCID:PMC7648733.
Sela TC, Iflah M, Muhsen K, Zahavi A. Descemet membrane endothelial keratoplasty compared with ultrathin Descemet stripping automated endothelial keratoplasty: a meta-analysis. BMJ open ophthalmology. 2023;8(1). doi:10.1136/bmjophth-2023-001397. PMID:37914389; PMCID:PMC10626808.
Dunker SL, Dickman MM, Wisse RPL, Nobacht S, Wijdh RHJ, Bartels MC, et al. Descemet Membrane Endothelial Keratoplasty versus Ultrathin Descemet Stripping Automated Endothelial Keratoplasty: A Multicenter Randomized Controlled Clinical Trial. Ophthalmology. 2020;127(9):1152-1159. doi:10.1016/j.ophtha.2020.02.029. PMID:32386811.
Krachmer JH, Purcell JJ Jr, Young CW, Bucher KD. Corneal endothelial dystrophy. A study of 64 families. Arch Ophthalmol. 1978;96(11):2036-2039. doi:10.1001/archopht.1978.03910060424004.
Gain P, Jullienne R, He Z, Aldossary M, Acquart S, Cognasse F, et al. Global Survey of Corneal Transplantation and Eye Banking. JAMA ophthalmology. 2016;134(2):167-73. doi:10.1001/jamaophthalmol.2015.4776. PMID:26633035.
Higa A, Sakai H, Sawaguchi S, Iwase A, Tomidokoro A, Amano S, Araie M.. Prevalence of and risk factors for cornea guttata in a population-based study in a southwestern island of Japan: the Kumejima study. Arch Ophthalmol. 2011;129(3):332-336. doi:10.1001/archophthalmol.2010.372. PMID:21402991.
Nakano M, Okumura N, Nakagawa H, Koizumi N, Ikeda Y, Ueno M, et al. Trinucleotide Repeat Expansion in the TCF4 Gene in Fuchs’ Endothelial Corneal Dystrophy in Japanese. Investigative ophthalmology & visual science. 2015;56(8):4865-9. doi:10.1167/iovs.15-17082. PMID:26218914.
Eric D. Wieben, Ross A. Aleff, Nirubol Tosakulwong, Malinda L. Butz, W. Edward Highsmith, Albert O. Edwards, Keith H. Baratz. A Common Trinucleotide Repeat Expansion within the Transcription Factor 4 (TCF4, E2-2) Gene Predicts Fuchs Corneal Dystrophy. PLoS ONE. 2012;7(11):e49083. doi:10.1371/journal.pone.0049083.
Zoega GM, Fujisawa A, Sasaki H, Kubota A, Sasaki K, Kitagawa K, et al. Prevalence and risk factors for cornea guttata in the Reykjavik Eye Study. Ophthalmology. 2006;113(4):565-9. doi:10.1016/j.ophtha.2005.12.014. PMID:16581419.
Melles GR, Ong TS, Ververs B, van der Wees J. Descemet membrane endothelial keratoplasty (DMEK). Cornea. 2006;25(8):987-990. doi:10.1097/01.ico.0001169600.69493.55.
Kinoshita S, Koizumi N, Ueno M, Okumura N, Imai K, Tanaka H, et al. Injection of Cultured Cells with a ROCK Inhibitor for Bullous Keratopathy. The New England journal of medicine. 2018;378(11):995-1003. doi:10.1056/NEJMoa1712770. PMID:29539291.
Moloney G, Petsoglou C, Ball M, Kerdraon Y, Höllhumer R, Spiteri N, et al. Descemetorhexis Without Grafting for Fuchs Endothelial Dystrophy-Supplementation With Topical Ripasudil. Cornea. 2017;36(6):642-648. doi:10.1097/ICO.0000000000001209. PMID:28476048.
Weiss JS, Møller HU, Aldave AJ, et al. IC3D classification of corneal dystrophies-Edition 2. Cornea. 2015;34(2):117-159. doi:10.1097/ICO.0000000000000307. PMID:25564336.
Seitzman GD, Gottsch JD, Stark WJ. Cataract surgery in patients with Fuchs’ corneal dystrophy: expanding recommendations for cataract surgery without simultaneous keratoplasty. Ophthalmology. 2005;112(3):441-446. doi:10.1016/j.ophtha.2004.10.044. PMID: 15745771.
Kopieren Sie den Artikeltext und fügen Sie ihn in den KI-Assistenten Ihrer Wahl ein.
Artikel in die Zwischenablage kopiert
Öffnen Sie unten einen KI-Assistenten und fügen Sie den kopierten Text in den Chat ein.