Grade 0-1(無至輕度)
所見:中央部12個或以下(Grade 0)或超過12個非融合性滴狀贅疣(Grade 1)
症狀:通常無症狀。鏡面顯微鏡下檢測為暗點。

Fuchs角膜內皮營養不良(FECD)是一種雙眼性角膜內皮細胞異常的進行性疾病。1910年,Ernst Fuchs首次將13例病例報告為「dystrophia epithelialis corneae」,後來被確認為內皮疾病,並得名1)。
角膜中央內皮面出現滴狀角膜(guttae/guttata),並逐漸向周邊擴散。當內皮細胞的屏障和幫浦功能(Na⁺/K⁺-ATP酶)下降時,會出現角膜基質水腫,進而發展為上皮水腫和水疱形成。Descemet膜增厚和不規則導致角膜透明性喪失。
在IC3D(國際角膜營養不良分類)第2版(Weiss 2015)中,FECD被歸類為「角膜內皮營養不良」類別15)。根據發病年齡大致分為以下兩型。
| 指標 | 數值 | 來源 |
|---|---|---|
| 白內障術前患者滴狀角膜發生率 | 1.2% | 國內多中心調查 |
| 日本Kumejima研究40歲以上盛行率 | 4.1% | Higa 20117) |
| 日本女性盛行率(40歲以上) | 5.8% | Higa 20117) |
| 日本:男性盛行率(40歲以上) | 2.4% | Higa 20117) |
| 冰島:雷克雅維克眼研究,55歲以上 | 女性11%,男性7% | Zoega 200610) |
| 性別比(國際) | 2.5:1至3.5:1(女性居多) | Matthaei 20191) |
| 日本人中TCF4重複擴增的頻率 | 47例中12例(26%) | Nakano 20158) |
身為黃種人的日本人,與白種人和黑種人相比,FECD的發生率往往較低。然而,隨著日本社會高齡化,預計未來病例數會進一步增加。日本人的角膜內皮細胞密度高於白人,因此被認為發病相對較晚。
在常伴有窄前房角的日本人中,雷射虹膜切開術(LI)後內皮細胞減少的病例並不少見,因此早期發現FECD需要特別注意。
在沖繩久米島進行的居民研究(Kumejima Study)中,40歲以上者有4.1%檢測到角膜滴狀贅疣(corneal guttae)。女性為5.8%,男性為2.4%7)。國內數據也顯示,接受白內障術前檢查的患者中有1.2%出現滴狀角膜。據報導,日本人發生率低於歐美人,但隨著高齡化社會的發展,發生率有增加趨勢。
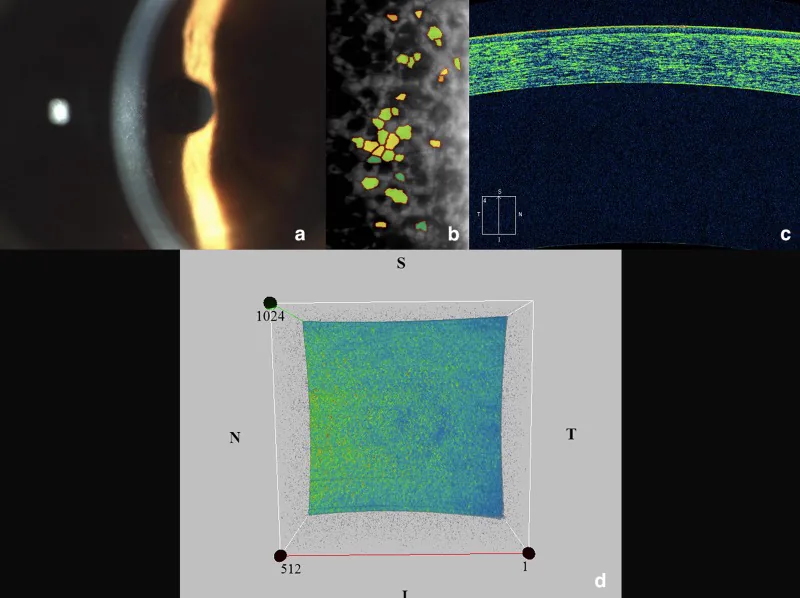
通常,50歲以下患者無症狀。症狀隨水腫程度緩慢進展。
裂隙燈顯微鏡檢查是基礎。結合直接照明、後方散射照明和鏡面反射法進行觀察。
Grade 0-1(無至輕度)
所見:中央部12個或以下(Grade 0)或超過12個非融合性滴狀贅疣(Grade 1)
症狀:通常無症狀。鏡面顯微鏡下檢測為暗點。
2-3級(中度)
所見: 中央1-5毫米融合性滴狀角膜。輕度錘擊金屬樣外觀。
症狀: 早晨視力模糊。鏡面顯微鏡下內皮影像不清晰。
4級(重度)
所見: 中央超過5毫米廣泛融合性滴狀角膜。伴有色素沉著的錘擊金屬樣外觀。
症狀: 從早晨到日間持續視力模糊和畏光。
4級+水腫(極重度)
所見: 基質水腫、上皮水腫和水疱形成。角膜明顯混濁。
症狀: 全天嚴重視力下降、眼痛和流淚。生活品質顯著下降。
該臨床分期基於Krachmer等人(1978年)的改良分類5)。
裂隙燈檢查所見細節:
健康的角膜透過內皮細胞不斷將水分泵入前房來保持透明。在FECD中,由於內皮泵功能下降,睡眠時(閉眼時)水分蒸發也停止,早晨角膜水腫最嚴重,霧視加重。睜眼時角膜表面水分蒸發,白天水腫有所改善,視力恢復。隨著疾病進展,這種日間波動消失,全天持續霧視。
FECD主要為體染色體顯性遺傳,但外顯率和表現度存在變異,部分病例無明確家族史。
主要致病基因:
在日本患者中,TCF4 重複擴增的頻率低於歐美人群,因此需要闡明其他遺傳背景8)。
FECD主要為體染色體顯性遺傳。理論上,遺傳給孩子的機率為50%。但發病年齡和嚴重程度差異很大(不完全外顯率),許多攜帶基因者終身僅有極輕微症狀。特別是,日本人中TCF4基因異常的比例低於西方國家8),遺傳背景可能不同。如有疑慮,建議諮詢遺傳專科醫師。
日本尚無統一的診斷標準,但透過以下檢查組合進行臨床診斷。
這是診斷和追蹤FECD最重要的檢查。
| 參數 | 正常值 | 異常閾值 |
|---|---|---|
| 內皮細胞密度(新生兒期) | 3,500~4,000 cells/mm² | — |
| 內皮細胞密度(20多歲) | 2,700 cells/mm² | — |
| 內皮細胞密度(70歲以上) | 平均約 2,200 個細胞/mm² | — |
| 維持透明性的最低密度 | — | 400~500 cells/mm² 以下 |
| CV值(變異係數) | 0.2~0.3 | ≥ 0.35 |
| 六角形細胞出現率(hexagonality) | 60〜70% | ≤ 50% |
| 疾病 | 鑑別要點 |
|---|---|
| 後部多形性角膜失養症(PPCD) | AD遺傳,雙眼性,Descemet膜帶狀/水泡樣混濁。基因:PPCD1(20p11.2-q11.2)、PPCD2(COL8A2)、PPCD3(ZEB1) |
| 先天性遺傳性角膜內皮失養症(CHED) | AR遺傳(SLC4A11突變),出生至嬰兒期發病,出生時即有角膜水腫和混濁 |
| 人工水晶體性水泡性角膜病變(PBK) | 白內障術後內皮損傷。無guttae,有手術史 |
| 假性剝落症候群角膜病變 | PEX物質沉積,眼壓升高,水晶體前囊PEX物質是鑑別關鍵 |
| 虹膜角膜內皮(ICE)症候群 | 單眼性,伴有虹膜萎縮、前粘連和青光眼。無guttae |
| 窄角眼的角膜內皮變化 | 可能出現滴狀角膜樣表現。透過眼壓和隅角形態鑑別 |
鏡面顯微鏡(鏡面反射型內皮細胞攝影裝置)是一種利用特殊光反射非侵入性拍攝和測量角膜最內層內皮細胞的設備。檢查測量內皮細胞數量(細胞密度)、大小變異(CV值)和形狀均勻性(六角形細胞比例)。在FECD中,guttae表現為黑點(暗點),有助於評估疾病分期。拍攝只需幾分鐘,無痛。
治療目標是恢復角膜透明度和維持視力。根據病期選擇對症治療或手術治療。
旨在手術前緩解症狀。不能恢復內皮細胞數量或抑制病情進展。
DMEK(後彈力層角膜內皮移植術)
移植物:僅後彈力層+內皮(厚度約15 μm)
特點:2006年Melles首次報告11)。視力恢復快,排斥率低。需要熟練的手術者。
日本保險適用:2016年起
DSAEK(後彈力層剝離自動角膜內皮移植術)
PKP(全層角膜移植)
移植片: 全層角膜(直徑7.0–8.5 mm)
特點: 經典選擇。縫合、散光管理及長期排斥風險為挑戰。在FECD領域,正逐漸被內皮移植術取代。
DSO(不聯合內皮移植的Descemet膜剝離術)
操作: 僅選擇性剝離中央4 mm的Descemet膜。無需移植片。
適應症: 殘留周邊內皮細胞能向中央遷移和增殖的病例。約75%可達角膜透明化14)。
ROCK抑制劑眼藥水: 術後併用利帕舒地爾可促進無反應病例的透明化14)。
| 指標 | DMEK | UT-DSAEK | 出處 |
|---|---|---|---|
| 12個月BCVA(logMAR差值) | −0.06(DMEK優勢) | — | Sela 2023統合分析3) |
| 20/25以上達成率 | 66% | 33%(p=0.02) | Dunker 2020 RCT4) |
| 再次氣泡注入的OR | — | 2.76(DSAEK優勢) | Sela 20233) |
| 12個月ECD | 無差異 | 無差異 | Dunker 20204) |
| 植片厚度<70 μm | — | 與DMEK視力無差異 | Sela 20233) |
Sela等人(2023年)的統合分析(8項研究,376隻眼)顯示,12個月時的BCVA在DMEK組顯著較佳(−0.06 logMAR)3)。Dunker等人(2020年)的多中心RCT也顯示,DMEK達到20/25或以上的比例高於UT-DSAEK(66% vs 33%,p=0.02)4)。然而,對於移植片厚度小於70 μm的UT-DSAEK,與DMEK的差異縮小了3)。
京都大學研究團隊(Kinoshita 2018)開發了一種治療方法,將培養的健康捐贈者角膜內皮細胞與ROCK抑制劑(Y-27632)同時注入前房13)。
ROCK抑制劑通過促進內皮細胞黏附、抑制凋亡和促進細胞週期進展來發揮作用13)。
FECD常合併白內障,需要謹慎選擇手術時機和方法。
正常角膜內皮細胞在前房內不會進行細胞分裂。當內皮缺損時,鄰近細胞會擴大並遷移以覆蓋缺損區域,因此細胞密度隨年齡增長而不可逆地下降。當低於400-500 cells/mm²時,維持角膜透明度變得困難。
在FECD中,變性的內皮細胞在Descemet膜後表面產生並沉積異常的膠原樣物質,形成guttae。Descemet膜增厚且不規則,形成進一步損害內皮功能的惡性循環。
老化、紫外線暴露和吸菸都會增加氧化壓力,成為惡性循環的入口2)。
角膜內皮的幫浦功能依賴於Na⁺/K⁺-ATP酶。當內皮細胞受損時,會經由以下途徑發生水腫。
如果眼壓升高(高眼壓)超過角膜基質的膨脹壓,即使內皮相對健康也可能發生上皮水腫,需注意。
由於TCF4重複擴增在日本人群中的貢獻相對較小8),闡明日本人特有的遺傳和環境背景是未來的重要課題。
Matthaei M, Hribek A, Clahsen T, Bachmann B, Cursiefen C, Jun AS. Fuchs Endothelial Corneal Dystrophy: Clinical, Genetic, Pathophysiologic, and Therapeutic Aspects. Annual review of vision science. 2019;5:151-175. doi:10.1146/annurev-vision-091718-014852. PMID:31525145.
Ong Tone S, Kocaba V, Böhm M, Wylegala A, White TL, Jurkunas UV. Fuchs endothelial corneal dystrophy: The vicious cycle of Fuchs pathogenesis. Progress in retinal and eye research. 2021;80:100863. doi:10.1016/j.preteyeres.2020.100863. PMID:32438095; PMCID:PMC7648733.
Sela TC, Iflah M, Muhsen K, Zahavi A. Descemet membrane endothelial keratoplasty compared with ultrathin Descemet stripping automated endothelial keratoplasty: a meta-analysis. BMJ open ophthalmology. 2023;8(1). doi:10.1136/bmjophth-2023-001397. PMID:37914389; PMCID:PMC10626808.
Dunker SL, Dickman MM, Wisse RPL, Nobacht S, Wijdh RHJ, Bartels MC, et al. Descemet Membrane Endothelial Keratoplasty versus Ultrathin Descemet Stripping Automated Endothelial Keratoplasty: A Multicenter Randomized Controlled Clinical Trial. Ophthalmology. 2020;127(9):1152-1159. doi:10.1016/j.ophtha.2020.02.029. PMID:32386811.
Krachmer JH, Purcell JJ Jr, Young CW, Bucher KD. Corneal endothelial dystrophy. A study of 64 families. Arch Ophthalmol. 1978;96(11):2036-2039. doi:10.1001/archopht.1978.03910060424004.
Gain P, Jullienne R, He Z, Aldossary M, Acquart S, Cognasse F, et al. Global Survey of Corneal Transplantation and Eye Banking. JAMA ophthalmology. 2016;134(2):167-73. doi:10.1001/jamaophthalmol.2015.4776. PMID:26633035.
Higa A, Sakai H, Sawaguchi S, Iwase A, Tomidokoro A, Amano S, Araie M.. Prevalence of and risk factors for cornea guttata in a population-based study in a southwestern island of Japan: the Kumejima study. Arch Ophthalmol. 2011;129(3):332-336. doi:10.1001/archophthalmol.2010.372. PMID:21402991.
Nakano M, Okumura N, Nakagawa H, Koizumi N, Ikeda Y, Ueno M, et al. Trinucleotide Repeat Expansion in the TCF4 Gene in Fuchs’ Endothelial Corneal Dystrophy in Japanese. Investigative ophthalmology & visual science. 2015;56(8):4865-9. doi:10.1167/iovs.15-17082. PMID:26218914.
Eric D. Wieben, Ross A. Aleff, Nirubol Tosakulwong, Malinda L. Butz, W. Edward Highsmith, Albert O. Edwards, Keith H. Baratz. A Common Trinucleotide Repeat Expansion within the Transcription Factor 4 (TCF4, E2-2) Gene Predicts Fuchs Corneal Dystrophy. PLoS ONE. 2012;7(11):e49083. doi:10.1371/journal.pone.0049083.
Zoega GM, Fujisawa A, Sasaki H, Kubota A, Sasaki K, Kitagawa K, et al. Prevalence and risk factors for cornea guttata in the Reykjavik Eye Study. Ophthalmology. 2006;113(4):565-9. doi:10.1016/j.ophtha.2005.12.014. PMID:16581419.
Melles GR, Ong TS, Ververs B, van der Wees J. Descemet membrane endothelial keratoplasty (DMEK). Cornea. 2006;25(8):987-990. doi:10.1097/01.ico.0001169600.69493.55.
Price MO, Feng MT, Price FW Jr. Endothelial Keratoplasty Update 2020. Cornea. 2021;40(5):541-547. doi:10.1097/ICO.0000000000002565. PMID:33252380.
Kinoshita S, Koizumi N, Ueno M, Okumura N, Imai K, Tanaka H, et al. Injection of Cultured Cells with a ROCK Inhibitor for Bullous Keratopathy. The New England journal of medicine. 2018;378(11):995-1003. doi:10.1056/NEJMoa1712770. PMID:29539291.
Moloney G, Petsoglou C, Ball M, Kerdraon Y, Höllhumer R, Spiteri N, et al. Descemetorhexis Without Grafting for Fuchs Endothelial Dystrophy-Supplementation With Topical Ripasudil. Cornea. 2017;36(6):642-648. doi:10.1097/ICO.0000000000001209. PMID:28476048.
Weiss JS, Møller HU, Aldave AJ, et al. IC3D classification of corneal dystrophies-Edition 2. Cornea. 2015;34(2):117-159. doi:10.1097/ICO.0000000000000307. PMID:25564336.
Seitzman GD, Gottsch JD, Stark WJ. Cataract surgery in patients with Fuchs’ corneal dystrophy: expanding recommendations for cataract surgery without simultaneous keratoplasty. Ophthalmology. 2005;112(3):441-446. doi:10.1016/j.ophtha.2004.10.044. PMID: 15745771.