Leber congenital amaurosis (LCA) is the most severe form of congenital retinal dystrophy, causing severe visual impairment from birth to infancy. It is a major cause of childhood visual impairment and is known as a representative cause of congenital visual impairment. Clinical manifestations are diverse.
It was first reported in 1869 by the German ophthalmologist Theodor Karl Gustav von Leber (1840–1917). Note that Leber hereditary optic neuropathy (LHON), reported by the same Leber in 1871, is a mitochondrial disease that develops around age 20 and is completely different from LCA. In 1957, the absence of waveforms on electroretinography (ERG) was confirmed as a common feature of LCA diagnosis, establishing the disease name.
The estimated birth prevalence is 2–3 per 100,000 births (1/30,000 to 1/81,000)1). Some reports cite 1:80,000 to 1:200,000, showing variability2). LCA accounts for about 5% of all retinal dystrophies, and about 20% of visually impaired children attending schools for the blind have LCA1). Currently, about 27 LCA-associated genes have been identified2), and causative genes are identified in about 70–80% of cases2). The inheritance pattern is mainly autosomal recessive, but autosomal dominant and X-linked inheritance have also been reported.
QWhen is Leber congenital amaurosis usually detected?
A
Parents often notice nystagmus or lack of fixation around 6 weeks of age3). When severe poor visual response (complete inability to fixate or follow) is observed, LCA is suspected and confirmed by electroretinography.
Higa N, et al. A novel RPE65 variant p.(Ala391Asp) in Leber congenital amaurosis: a case report and literature review in Japan. Front Med (Lausanne). 2024. Figure 1. PMCID: PMC11446184. License: CC BY.
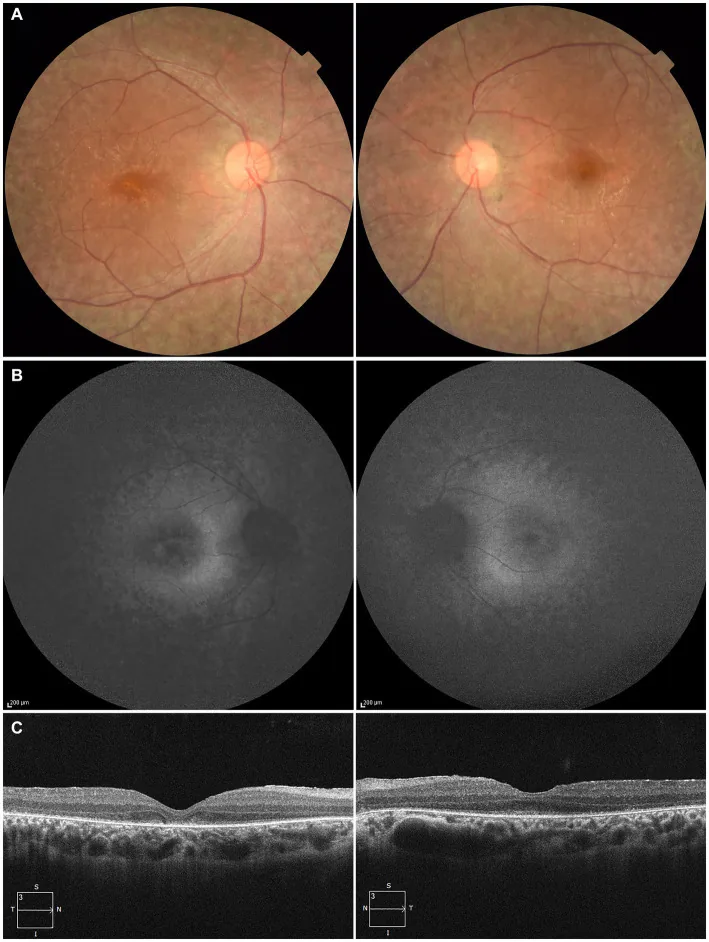
A 27-year-old patient: (A) Fundus photographs of both eyes showing retinal pigment epithelium depigmentation and retinal vascular attenuation; (B) Fundus autofluorescence showing hyperautofluorescence and signal loss in affected areas; (C) OCT showing loss of the ellipsoid zone (EZ) in the right eye and minimal residual EZ at the fovea in the left eye. Corresponds to retinal pigment epithelium degeneration and retinal vascular attenuation discussed in section “2. Main symptoms and clinical findings”.
Severe visual impairment is present from birth or early infancy.
Visual acuity loss: Most patients have visual acuity of 0.1 or less, and about one-third have no light perception. Visual impairment is generally stable or progresses very slowly.
Photophobia: Many patients show sensitivity to light.
Night blindness: Visual function in dark conditions is further reduced.
Parents often notice nystagmus or lack of fixation around 6 weeks of age 3).
Clinical findings (findings confirmed by physician examination)
Nystagmus: Appears at birth or soon after. It is pendular or wandering and present in all gaze positions. This is an important distinguishing feature from early-onset severe retinal dystrophy (EOSRD), which does not involve nystagmus2).
Abnormal pupillary response: The pupillary light reflex is sluggish or absent. This is called “amaurotic pupils.”
Oculo-digital sign: Poking, pressing, or rubbing the eyes, thought to be an attempt to mechanically stimulate the retina to elicit visual sensations. The main sequela is enophthalmos due to orbital fat atrophy.
Refractive error: High hyperopia (>5 diopters) is common, thought to result from impaired emmetropization due to early visual impairment.
In infancy, the fundus often appears normal, but various findings appear later. Fundus findings range from normal to typical retinitis pigmentosa-like appearance. In advanced cases, the optic disc becomes pale, blood vessels become extremely attenuated, the overall fundus color darkens, and the foveal reflex disappears.
Optic disc pallor and retinal vascular attenuation
Salt-and-pepper fundus: retinal pigment epithelium degeneration with small white spots and depigmentation
Bone-spicule pigmentation and subretinal flecks (marbled fundus)
On OCT, the outer retinal layers are almost absent, with thinning to loss of the ellipsoid zone (IS/OS junction) being characteristic. Findings vary by genotype:
CRB1 mutation: paradoxical finding of increased retinal thickness (coarse lamination)1)
QWhat is the purpose of the oculo-digital sign (eye rubbing)?
A
This is a characteristic behavior in LCA patients, thought to mechanically stimulate the retina by poking or pressing the eyes with fingers, attempting to evoke visual sensations. Repeated over time, it leads to atrophy of orbital fat and causes enophthalmos.
LCA is a group of inherited retinal degenerative diseases, most of which follow an autosomal recessive inheritance pattern2). Rarely, mutations in CRX, IMPDH1, or OTX2 show autosomal dominant inheritance2). X-linked inheritance has also been reported.
Currently, 19 types (LCA1 to LCA19) and 8 additional related genes have been reported2). The causative genes are involved in multiple pathways related to retinal development and function, including photoreceptor morphogenesis, phototransduction cilia, and the visual cycle.
LCA-associated genes are classified into five major functional networks2):
Retinoid metabolism / rod visual cycle (RPE65, LRAT, RDH12)
The most frequently mutated genes worldwide and their proportions are as follows:
Gene
Proportion
Pathway involved
CEP290
Approximately 15%
Ciliary function
GUCY2D
Approximately 12%
Phototransduction (cGMP synthesis)
CRB1
Approximately 10%
Cell polarity maintenance
RPE65
Approximately 8%
Retinoid metabolism
In a Japanese cohort (34 families), NGS analysis showed a detection rate of about 56%, with the most frequent mutated genes reported as CRB1, NMNAT1, and RPGRIP11).
Identification of the causative gene directly determines treatment eligibility. In particular, confirmation of RPE65 mutation is essential for determining eligibility for gene therapy (voretigene neparvovec)2).
AIPL1 functions as a specific chaperone for phosphodiesterase 6 (PDE6), which is responsible for cGMP degradation in phototransduction. AIPL1 deficiency leads to a dramatic reduction in PDE6 protein levels, resulting in cGMP metabolic disruption, photoreceptor degeneration, and early blindness2). AIPL1 mutations account for approximately 5–10% of all LCA cases2).
QWhat is the probability that the next child will also have LCA?
A
In autosomal recessive inheritance, if both parents are carriers, the probability that the next child will be affected is 25%, the probability of being a carrier is 50%, and the probability of being unaffected and non-carrier is 25%. If the causative gene is identified, prenatal diagnosis or preimplantation genetic diagnosis are options.
The diagnosis of LCA is made clinically, and confirmation requires electroretinography for definitive diagnosis and genetic testing for molecular genetic confirmation2).
LCA is suspected when there is a congenital marked lack of visual response (lack of fixation and tracking). In infants with severe visual impairment and high hyperopia, molecular genetic testing for LCA is the first choice4).
Varies by subtype (absent in RPE65 type, normal in GUCY2D type).
Genetic testing (NGS, etc.)
Required for definitive diagnosis and subtype identification.
Electroretinogram: Both rod and cone responses are absent to severely reduced. A normal ERG rules out LCA.
OCT: The outer retinal layers and ellipsoid zone are mostly absent. CRB1 mutations show paradoxical retinal thickening (coarse lamination) 1). Handheld OCT is useful for examining awake infants or young children under anesthesia 4).
FAF: Findings vary by subtype. GUCY2D mutations show preserved autofluorescence, while RPE65 mutations show absent autofluorescence 2).
Genetic testing: Next-generation sequencing (NGS), DNA microarrays, and linkage analysis are used. The overall detection rate is approximately 70–80% 2). Since 2023, a panel test for 82 IRD causative genes (PrismGuide IRD Panel) has been covered by insurance and is applied to young-onset patients suspected of having RPE65-related IRD.
For most types of LCA, no substantial treatment has been established. Current management is as follows.
Correction of refractive errors: Provide appropriate refractive correction for high hyperopia, etc. Since strong refractive errors may be present, prescribe glasses and strive for vision training.
Vision training: Provide visual rehabilitation to maximize the use of residual visual function.
Vision substitution training: Because visual function is severely impaired, consider Braille instruction, white cane walking training, use of magnifying reading devices, etc.
In 2017, the US FDA approved voretigene neparvovec-rzyl (brand name Luxturna) as a treatment for LCA2 associated with biallelic RPE65 mutations. This is the first FDA-approved gene therapy product in ophthalmology. In 2023, it was also approved in Japan (product name: Luxturna® injection).
A recombinant adeno-associated virus (rAAV2) vector delivers a normal copy of the RPE65 gene to the retinal pigment epithelium via subretinal injection. The procedure is performed in a vitreous surgery operating room.
Phase III 301 trial (Russell 2017)5):
Subjects: 31 patients with RPE65-associated IRD
Primary endpoint: MLMT (multi-luminance mobility test; an observational test of mobility under various light levels)
Significant improvement was also observed in full-field stimulus threshold (FST)
Phase I/III long-term results (Maguire 2019)6):
Retinal sensitivity, visual acuity, and functional gains that peaked at 6–12 months after treatment showed a tendency to progressively decline thereafter
Improvement in night blindness and visual field can be expected due to increased retinal sensitivity
Domestic Phase III trial (A11301 trial) (Fujinami 2025)7):
Subjects: 4 Japanese patients with RPE65-associated IRD
Significant sensitivity increase in FST (full-field stimulus threshold) (defined as a sensitivity increase of more than 10-fold)
Visual field expansion was confirmed at 1 year after administration
Main adverse events: ocular disorders including eye pain (presumed to be related to the administration procedure)
Administration protocol:
The second eye is administered at least 6 days after the first eye.
Immunosuppression: Start steroids 3 days before administration and continue for 14 days after administration.
However, RPE65 mutations account for only about 8% of all LCA patients. For other mutation types, there is currently no proven treatment.
QCan gene therapy be used for all LCA patients?
A
Currently, the approved gene therapy (voretigene neparvovec / Luxturna® injection) is indicated only for LCA2 caused by biallelic RPE65 mutations. RPE65 mutations account for about 8% of all LCA cases, so the majority of patients are not eligible. Treatments for other genotypes are still in the research stage.
QHow can I receive gene therapy in Japan?
A
Eligibility requires confirmation of biallelic RPE65 mutations and sufficient viable retinal cells. Genetic diagnosis using a gene panel test (PrismGuide IRD panel) is the first step. Referral to a specialized facility with experience in treating inherited retinal diseases is recommended.
6. Pathophysiology and detailed mechanism of onset
The pathophysiology of LCA is related to the disruption of the visual cycle, which prevents the eye from transmitting light information.
The visual cycle is a series of enzymatic reactions between the retinal pigment epithelium (RPE) and the neurosensory retina, metabolizing dietary vitamin A to produce 11-cis retinal, which generates photopigments. Without 11-cis retinal, the phototransduction cascade cannot be initiated, and visual nerve signals are not transmitted to the visual cortex. Mutations in any of the genes encoding proteins involved in this cascade can block the visual cycle and cause LCA symptoms.
Histopathologically, involvement of the outer retina and photoreceptors has been demonstrated, suggesting that LCA is a degenerative process rather than a malformation.
GUCY2D (LCA1): Encodes retinal-specific guanylate cyclase (GC-E). Catalyzes cGMP synthesis and is key to phototransduction. Over 140 disease-associated mutations have been identified, 88% causing autosomal recessive LCA. Amino acid 838 is known as a mutation hotspot 2).
RPE65 (LCA2): Belongs to the carotenoid cleavage oxygenase superfamily. It is a bifunctional enzyme that catalyzes O-alkyl ester cleavage from all-trans-retinyl ester and isomerization of the retinyl moiety (all-trans-retinol → 11-cis-retinol) 2). Essential for both rod and cone function, and recent studies suggest it may also be involved in the isomerization of lutein to meso-zeaxanthin 2). It is the only approved indication for gene therapy.
CRB1 (LCA8): Homologous to the Drosophila crumbs protein, expressed in photoreceptor inner segments and Müller cells. Important for maintaining cell polarity, located on chromosome 1q31.3 1).
CEP290 (LCA10): Involved in ciliary function of photoreceptors. It has the highest mutation frequency among LCA-associated genes (approximately 15%).
NMNAT1 (LCA9): Encodes a key enzyme in NAD (nicotinamide adenine dinucleotide) biosynthesis 2).
LCA5: Encodes lebercilin, involved in ciliary function and intraflagellar transport 2).
AIPL1: Functions as a specialized chaperone for PDE6 (phosphodiesterase 6). AIPL1 deficiency leads to PDE6 instability → cGMP metabolism disruption → channel abnormalities → photoreceptor degeneration 2).
7. Latest research and future perspectives (reports at research stage)
Long-term follow-up evaluations of voretigene neparvovec (NCT00481546, NCT00643747) have reported that after an initial peak at 6–12 months post-treatment, clinical benefits including retinal sensitivity, visual acuity, and functional gains progressively decline 6). The PERCEIVE study (prospective registry study) reported 2-year safety and efficacy data in real-world clinical practice, with GTAU (gene therapy-associated uveitis) observed in up to 50% of cases 9).
Developed by Editas Medicine. An AAV5 vector carrying S. aureus-derived Cas9 and two guide RNAs targets the deep intronic mutation (c.2991+1655A>G) in intron 26 of CEP290 3). Safety was confirmed in a first-in-human trial, showing good tolerability even at relatively high doses 3).
As a gene-independent approach, a method to express light-responsive channelrhodopsin in remaining inner retinal neurons is being studied 3). It may be applicable to all LCA types (regardless of genotype), and early clinical trials are ongoing.
The course of LCA is classified into three patterns: stable (about 75%), progressive worsening (about 15%), and improvement (about 10%). AIPL1 mutations are associated with progressive worsening, while RPGRIP1 mutations are associated with a stable course. In the future, halting progression or treatment through artificial vision, gene therapy, and regenerative medicine is expected.
Duan W, Zhou T, Jiang H, Zhang M, Hu M, Zhang L. A novel nonsense variant (c.1499C>G) in CRB1 caused Leber congenital amaurosis-8 in a Chinese family and a literature review. BMC Med Genomics. 2022;15(1):197. doi:10.1186/s12920-022-01356-z.
Mordà D, Alibrandi S, Scimone C, Rinaldi C, Scalinci SZ, Abate G, et al. Decoding pediatric inherited retinal dystrophies: Bridging genetic complexity and clinical heterogeneity. Progress in retinal and eye research. 2025;109:101405. doi:10.1016/j.preteyeres.2025.101405. PMID:40998214.
Botto C, Rucli M, Tekinsoy MD, Pulman J, Sahel JA, Dalkara D. Early and late stage gene therapy interventions for inherited retinal degenerations. Progress in retinal and eye research. 2022;86:100975. doi:10.1016/j.preteyeres.2021.100975. PMID:34058340.
Gurnani B, et al. Clinical approach to pediatric nystagmus: a comprehensive diagnostic algorithm. Clin Ophthalmol. 2025;19:1617-1636.
Russell S, Bennett J, Wellman JA, Chung DC, Yu ZF, Tillman A, et al. Efficacy and safety of voretigene neparvovec (AAV2-hRPE65v2) in patients with RPE65-mediated inherited retinal dystrophy: a randomised, controlled, open-label, phase 3 trial. Lancet (London, England). 2017;390(10097):849-860. doi:10.1016/S0140-6736(17)31868-8. PMID:28712537; PMCID:PMC5726391.
Maguire AM, Russell S, Wellman JA, Chung DC, Yu ZF, Tillman A, et al. Efficacy, Safety, and Durability of Voretigene Neparvovec-rzyl in RPE65 Mutation-Associated Inherited Retinal Dystrophy: Results of Phase 1 and 3 Trials. Ophthalmology. 2019;126(9):1273-1285. doi:10.1016/j.ophtha.2019.06.017. PMID:31443789.
Fujinami K, Akiyama K, Tsunoda K, et al. Efficacy and Safety of Voretigene Neparvovec in RPE65-Retinopathy: Results of a Phase III Trial in Japan. Ophthalmol Sci. 2025;5:100876. doi:10.1016/j.xops.2025.100876. PMID:40910102; PMCID:PMC12405627.
Gange WS, Sisk RA, Besirli CG, Lee TC, Havunjian M, Schwartz H, et al. Perifoveal Chorioretinal Atrophy after Subretinal Voretigene Neparvovec-rzyl for RPE65-Mediated Leber Congenital Amaurosis. Ophthalmology. Retina. 2022;6(1):58-64. doi:10.1016/j.oret.2021.03.016. PMID:33838313; PMCID:PMC8497635.
Fischer MD, Simonelli F, Sahni J, Holz FG, Maier R, Fasser C, et al. Real-World Safety and Effectiveness of Voretigene Neparvovec: Results up to 2 Years from the Prospective, Registry-Based PERCEIVE Study. Biomolecules. 2024;14(1). doi:10.3390/biom14010122. PMID:38254722; PMCID:PMC10813228.
Copy the article text and paste it into your preferred AI assistant.
Article copied to clipboard
Open an AI assistant below and paste the copied text into the chat box.