A amaurose congênita de Leber (LCA) é a forma mais grave das distrofias retinianas congênitas, causando deficiência visual grave desde o nascimento até a infância. É uma das principais causas de deficiência visual infantil e é conhecida como causa comum de cegueira congênita. O quadro clínico é variado.
Foi relatada pela primeira vez em 1869 pelo oftalmologista alemão Theodor Karl Gustav von Leber (1840–1917). Note-se que a neuropatia óptica hereditária de Leber (LHON), relatada pelo mesmo Leber em 1871, é uma doença mitocondrial que surge por volta dos 20 anos e é completamente diferente da LCA. Em 1957, a ausência de ondas no ERG foi confirmada como característica comum no diagnóstico da LCA, estabelecendo o nome da doença.
A prevalência estimada ao nascimento é de 2 a 3 por 100.000 nascimentos (1/30.000 a 1/81.000)1). Alguns relatos mencionam 1:80.000 a 1:200.000, indicando variação2). A LCA representa cerca de 5% de todas as distrofias retinianas, e aproximadamente 20% das crianças com deficiência visual em escolas para cegos têm LCA1). Atualmente, cerca de 27 genes relacionados à LCA foram identificados2), e o gene causador é encontrado em cerca de 70–80% dos casos2). O padrão de herança principal é autossômico recessivo, mas há relatos de herança dominante e ligada ao X.
QQuando a amaurose congênita de Leber pode ser detectada?
A
Geralmente, os pais percebem nistagmo ou falta de fixação por volta das 6 semanas de idade3). Se houver resposta visual grave (incapacidade de fixar ou seguir), suspeita-se de LCA e o diagnóstico é confirmado por ERG.
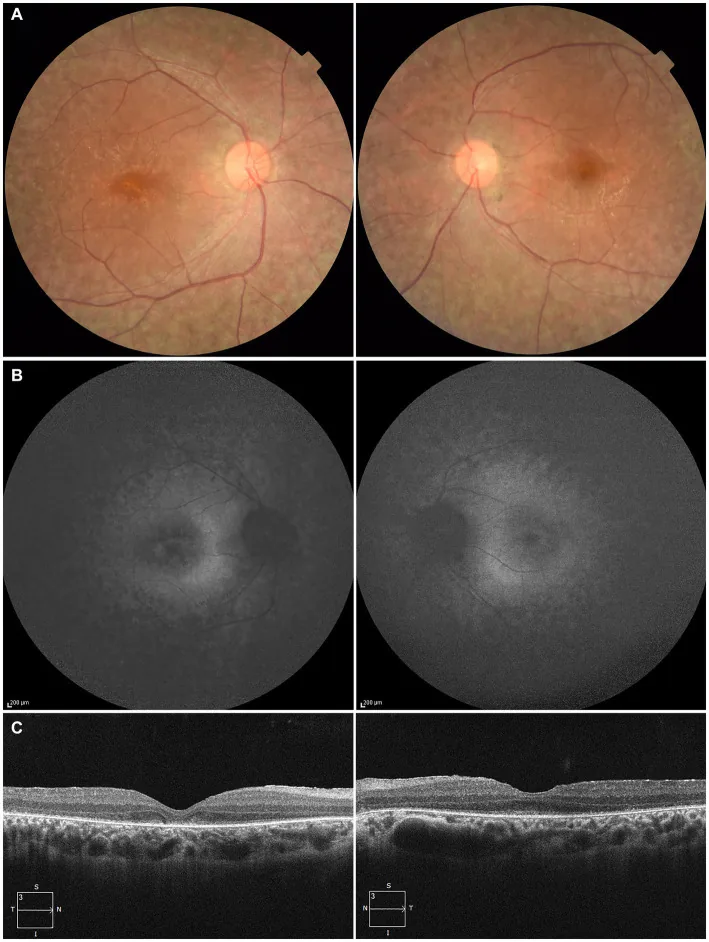
Higa N, et al. A novel RPE65 variant p.(Ala391Asp) in Leber congenital amaurosis: a case report and literature review in Japan. Front Med (Lausanne). 2024. Figure 1. PMCID: PMC11446184. License: CC BY.
Em paciente de 27 anos, (A) fotografia de fundo de ambos os olhos mostrando despigmentação do epitélio pigmentar da retina e estreitamento dos vasos retinianos, (B) autofluorescência de fundo mostrando hiperfluorescência e redução de sinal na lesão, (C) OCT mostrando desaparecimento da zona elipsoide (EZ) no olho direito e pequeno resquício na fóvea do olho esquerdo. Corresponde à degeneração do epitélio pigmentar da retina e estreitamento dos vasos retinianos discutidos na seção “2. Principais sintomas e achados clínicos”.
Deficiência visual grave está presente desde o nascimento ou início do período pós-natal.
Baixa acuidade visual: A maioria dos pacientes tem acuidade visual inferior a 0,1, e cerca de um terço não tem percepção de luz. A deficiência visual geralmente é estável ou progride muito lentamente.
Fotofobia: Muitos casos apresentam hipersensibilidade à luz.
Cegueira noturna: A função visual em ambientes escuros piora ainda mais.
Frequentemente, os pais percebem nistagmo ou falta de fixação por volta das 6 semanas de idade3).
Achados clínicos (achados confirmados pelo médico no exame)
Nistagmo: Aparece ao nascimento ou logo após. É pendular ou errante em todas as posições do olho. É um importante ponto de diferenciação com a distrofia retiniana grave de início precoce (EOSRD), na qual não há nistagmo2).
Anormalidade da reação pupilar: Reflexo fotomotor lento ou ausente. Chamado de “pupilas amauróticas (amaurotic pupils)”.
Sinal oculodigital (oculo-digital sign): Movimento de cutucar, pressionar ou esfregar os olhos, considerado uma tentativa de estimular mecanicamente a retina para evocar visão. A principal sequela é a enoftalmia devido à atrofia da gordura orbitária.
Erro refrativo: Hipermetropia alta (>5 dioptrias) é comum, acredita-se que seja devido à falha de emetropização causada pela deficiência visual precoce.
Na infância, o fundo de olho frequentemente parece normal, mas posteriormente surgem achados variados. Os achados de fundo variam de fundo normal a fundo típico de retinite pigmentosa. Em casos avançados, o disco óptico torna-se pálido, os vasos sanguíneos tornam-se extremamente estreitos, a coloração geral do fundo escurece e o reflexo anular macular desaparece.
Palidez do disco óptico e estreitamento dos vasos retinianos
Fundo de olho em sal e pimenta: degeneração do epitélio pigmentar da retina com pequenas manchas brancas e despigmentação
Pigmentação em espículas ósseas e flecks subretinianos (fundo marmóreo)
Degeneração macular e exsudação semelhante à doença de Coats
Na OCT, as camadas externas da retina estão quase ausentes, com afinamento a desaparecimento da zona elipsoide (junção IS/OS). Os achados variam conforme o genótipo:
Mutação CRB1: aumento paradoxal da espessura retiniana (laminação grosseira) 1)
Mutação RPE65: desaparecimento da FAF (autofluorescência do fundo) 2)
QQual é o propósito do sinal oculodigital (movimento de esfregar os olhos)?
A
Comportamento característico de pacientes com LCA, onde cutucam ou pressionam o olho com os dedos para estimular mecanicamente a retina e tentar evocar a visão. A repetição a longo prazo causa atrofia da gordura orbital, levando à enoftalmia.
LCA é um grupo de doenças hereditárias de degeneração retiniana, a maioria seguindo padrão de herança autossômica recessiva2). Raramente, casos autossômicos dominantes ocorrem devido a mutações em CRX, IMPDH1 e OTX22). Também há relatos de herança ligada ao X.
Atualmente, 19 tipos de LCA1 a LCA19 foram relatados, além de 8 genes relacionados adicionais2). Os genes causadores estão envolvidos em múltiplas vias relacionadas ao desenvolvimento e função da retina, como morfogênese dos fotorreceptores, cílios de transdução de luz e ciclo visual.
Os genes relacionados à LCA são classificados em 5 redes funcionais principais2):
Metabolismo de retinoides e ciclo visual dos bastonetes (RPE65, LRAT, RDH12)
Manutenção da homeostase da retina e manutenção dos fotorreceptores (AIPL1, SPATA7, TULP1, USP45, CRB1, LCA5)
Desenvolvimento da retina e morfogênese (RD3, CEP290)
Detecção de estímulo luminoso e percepção visual (GUCY2D, CNGA3)
Cílios conectores dos fotorreceptores e manutenção do segmento externo (CEP290, RPGRIP1, RPGR)
Os genes mais frequentemente mutados no mundo e suas proporções são os seguintes:
Gene
Proporção
Via envolvida
CEP290
Cerca de 15%
Função ciliar
GUCY2D
Cerca de 12%
Transdução de sinal luminoso (síntese de cGMP)
CRB1
Cerca de 10%
Manutenção da polaridade celular
RPE65
Cerca de 8%
Metabolismo de retinoides
Em uma coorte japonesa (34 famílias), a análise por NGS mostrou uma taxa de detecção de cerca de 56%, e os genes mais frequentemente mutados relatados foram CRB1, NMNAT1 e RPGRIP11).
A identificação do gene causador está diretamente ligada à determinação da elegibilidade para tratamento. Em particular, a confirmação da mutação RPE65 é essencial para a avaliação da elegibilidade para terapia gênica (voretigene neparvovec)2).
AIPL1 funciona como uma chaperona especializada para a fosfodiesterase 6 (PDE6), que degrada o cGMP na transdução do sinal luminoso. A deficiência de AIPL1 leva a uma redução drástica na quantidade da proteína PDE6, resultando em colapso do metabolismo do cGMP → degeneração dos fotorreceptores → cegueira precoce2). Mutações no AIPL1 representam cerca de 5-10% de todos os casos de LCA2).
QQual a probabilidade de o próximo filho herdar LCA?
A
Na herança autossômica recessiva, se ambos os pais são portadores, a probabilidade de o próximo filho ser afetado é de 25%, a probabilidade de ser portador é de 50% e a probabilidade de não ser afetado nem portador é de 25%. Se o gene causador for identificado, o diagnóstico pré-natal ou o diagnóstico pré-implantacional podem ser opções.
O diagnóstico de LCA é feito clinicamente, sendo necessária a confirmação por eletrorretinografia e confirmação genética molecular por teste genético2).
Quando há uma resposta visual congênita gravemente deficiente (falta de fixação e seguimento), suspeita-se de LCA. Em lactentes com deficiência visual grave e hipermetropia alta, a pesquisa de LCA por teste genético molecular é a primeira escolha4).
Os principais exames diagnósticos são mostrados abaixo.
Varia conforme o subtipo (tipo RPE65: ausente, tipo GUCY2D: normal).
Teste genético (NGS, etc.)
Necessário para diagnóstico definitivo e identificação do subtipo.
Eletrorretinografia: Respostas de bastonetes e cones ausentes ou acentuadamente reduzidas. ERG normal exclui o diagnóstico de LCA.
OCT: As camadas externas da retina e a zona elipsoide estão quase ausentes. Em mutações CRB1, observa-se um aumento paradoxal da espessura retiniana (laminação grosseira) 1). OCT portátil é útil para examinar lactentes acordados ou crianças pequenas sob anestesia 4).
FAF: Os achados variam conforme o subtipo. Em mutações GUCY2D, a autofluorescência é normal; em mutações RPE65, está ausente 2).
Teste genético: Utilizam-se sequenciamento de nova geração (NGS), microarranjo de DNA, análise de ligação. A taxa de detecção geral é de cerca de 70-80% 2). Desde 2023, um teste de painel para 82 genes causadores de IRD (Painel PrismGuide IRD) é coberto pelo seguro, aplicado a casos de início precoce com suspeita de IRD relacionada ao RPE65.
Não há tratamento substancial estabelecido para a maioria dos tipos de LCA. O manejo atual é o seguinte:
Correção de erros refrativos: Realizar correção refrativa adequada para hipermetropia alta, etc. Pode haver erro refrativo significativo, portanto prescrever óculos e buscar treinamento visual.
Treinamento visual: Realizar reabilitação visual para maximizar o uso da função visual residual.
Treinamento de substituição visual: Devido ao comprometimento significativo da função visual, considerar instrução em Braille, treinamento de mobilidade com bengala e uso de lupas de leitura.
Cuidados de baixa visão: Uso de auxílios para baixa visão, apoiar o acesso ideal a oportunidades educacionais e de emprego.
Aconselhamento genético: Recomendado para famílias e pacientes. Teste de portador, diagnóstico pré-natal e diagnóstico genético pré-implantacional podem ser possíveis.
Manejo da fotofobia: Recomenda-se o uso de óculos de proteção contra luz e redução da exposição à luz.
Acompanhamento regular: Realizar acompanhamento oftalmológico incluindo eletrorretinograma, encaminhar para ambulatório de baixa visão quando necessário.
Em 2017, o FDA dos EUA aprovou o voretigene neparvovec-rzyl (nome comercial Luxturna) como tratamento para LCA2 associado a mutações bialélicas do RPE65. Este foi o primeiro produto de terapia genética aprovado pelo FDA na área oftalmológica. Em 2023, também foi aprovado no Japão (nome do produto: Luxturna®注).
Uma cópia normal do gene RPE65 é introduzida no epitélio pigmentar da retina por injeção sub-retiniana usando um vetor de vírus adeno-associado recombinante (rAAV2). O procedimento é realizado em sala de cirurgia de vitrectomia.
Estudo de Fase III 301 (Russell 2017)5):
Participantes: 31 pacientes com IRD relacionado ao RPE65
Desfecho primário: MLMT (teste de mobilidade em múltiplas luminâncias; teste de observação comportamental em diferentes níveis de iluminação)
Melhora significativa também foi observada no limiar de estímulo de campo total (FST)
Resultados de longo prazo da Fase I/III (Maguire 2019)6):
A sensibilidade retiniana, acuidade visual e ganhos funcionais que atingiram o pico 6-12 meses após o tratamento mostraram uma tendência de declínio progressivo posteriormente
O aumento da sensibilidade retiniana pode melhorar a cegueira noturna e o campo visual
Estudo de Fase III nacional (estudo A11301) (Fujinami 2025)7):
Participantes: 4 pacientes japoneses com IRD relacionado ao RPE65
Aumento significativo da sensibilidade no FST (limiar de estímulo de campo total) (aumento de sensibilidade superior a 10 vezes foi definido como significativo)
A expansão do campo visual foi confirmada 1 ano após a administração
Principais eventos adversos: distúrbios oculares incluindo dor ocular (presumivelmente relacionados ao procedimento de administração)
Protocolo de administração:
O segundo olho é administrado pelo menos 6 dias após o primeiro
Imunossupressão: iniciar corticosteroide 3 dias antes da administração e continuar por 14 dias após
No entanto, mutações no RPE65 representam apenas cerca de 8% de todos os pacientes com LCA. Para outros tipos de mutação, atualmente não há terapia com eficácia comprovada.
QA terapia gênica pode ser usada em todos os pacientes com LCA?
A
A terapia gênica atualmente aprovada (voretigene neparvovec / Luxturna®) é indicada apenas para LCA2 causada por mutações bialélicas no RPE65. Mutações no RPE65 representam apenas cerca de 8% de todos os casos de LCA, portanto a maioria dos pacientes não é elegível. Terapias para outros genótipos ainda estão em fase de pesquisa.
QComo receber terapia gênica no Japão?
A
É necessário confirmar mutações bialélicas no RPE65 e ter células retinianas viáveis suficientes. O primeiro passo é o diagnóstico genético por meio de um painel genético (PrismGuide IRD panel). Recomenda-se o encaminhamento para centros especializados com experiência em doenças retinianas hereditárias.
6. Fisiopatologia e mecanismo detalhado de ocorrência
A fisiopatologia da LCA está relacionada à interrupção do ciclo visual (Visual Cycle), impedindo que o olho transmita informações luminosas.
O ciclo visual é uma série de reações enzimáticas entre o epitélio pigmentar da retina (EPR) e a retina neurosensorial, no qual a vitamina A da dieta é metabolizada para produzir 11-cis-retinal, necessário para formar os pigmentos visuais. Sem o 11-cis-retinal, a cascata de transdução do sinal luminoso não pode ser iniciada e os sinais nervosos visuais não são transmitidos ao córtex visual. Mutações em qualquer um dos genes que codificam proteínas envolvidas nessa série de reações interrompem o ciclo visual e causam os sintomas da LCA.
Histopatologicamente, há evidências de envolvimento da retina externa e dos fotorreceptores, sugerindo que a LCA é um processo degenerativo, e não displásico.
GUCY2D (LCA1): Codifica a guanilato ciclase específica da retina (GC-E). Catalisa a síntese de cGMP, chave na transdução de sinal luminoso. Mais de 140 mutações associadas à doença foram identificadas, 88% causam LCA autossômica recessiva. O aminoácido 838 é conhecido como ponto quente de mutação 2).
RPE65 (LCA2): Pertence à superfamília das carotenoid cleavage oxigenases, é uma enzima bifuncional que catalisa a clivagem da ligação O-alquil do all-trans-retinyl ester e a isomerização do grupo retinil (all-trans-retinol → 11-cis-retinol) 2). Essencial para a função de bastonetes e cones, estudos recentes sugerem possível envolvimento na isomerização de luteína em meso-zeaxantina 2). Única terapia gênica aprovada.
CRB1 (LCA8): Homólogo à proteína crumbs da Drosophila, expresso no segmento interno dos fotorreceptores e nas células de Müller. Importante para a manutenção da polaridade celular, localizado no cromossomo 1q31.3 1).
CEP290 (LCA10): Envolvido na função ciliar dos fotorreceptores. Maior frequência de mutação entre os genes relacionados à LCA (cerca de 15%).
NMNAT1 (LCA9): Codifica a enzima chave na biossíntese de NAD (nicotinamida adenina dinucleotídeo) 2).
LCA5: Codifica a lebercilina, envolvida na função ciliar e no transporte de proteínas intraciliares 2).
AIPL1: Funciona como chaperona especializada para PDE6 (fosfodiesterase 6). A deficiência de AIPL1 leva à instabilidade da PDE6 → ruptura do metabolismo de cGMP → anormalidade dos canais → degeneração fotorreceptora 2).
7. Pesquisas recentes e perspectivas futuras (relatos em fase de pesquisa)
Na avaliação de longo prazo do voretigene neparvovec (NCT00481546, NCT00643747), foi relatado um pico inicial 6-12 meses após o tratamento, seguido por declínio progressivo dos benefícios clínicos, incluindo sensibilidade retiniana, acuidade visual e ganho funcional 6). O estudo PERCEIVE (estudo prospectivo de registro) relatou dados de segurança e eficácia de 2 anos na prática clínica real, com uveíte associada à terapia gênica (GTAU) observada em até 50% dos casos 9).
Desenvolvido pela Editas Medicine. O vetor AAV5 carrega Cas9 de S. aureus e dois RNAs guia, visando a mutação intrônica profunda (c.2991+1655A>G) localizada no íntron 26 do CEP290 3). O primeiro estudo em humanos confirmou segurança e mostrou boa tolerabilidade mesmo em doses relativamente altas 3).
Como abordagem independente de gene, a técnica de expressar canalrodopsina responsiva à luz em neurônios retinianos internos remanescentes está sendo estudada 3). Potencialmente aplicável a todos os tipos de LCA (independentemente do genótipo), e ensaios clínicos iniciais estão em andamento.
A terapia gênica para mutações GUCY2D e AIPL1 está em andamento em modelos animais e mostrou resultados promissores no resgate de fotorreceptores bastonetes e cones.
O curso da LCA é classificado em três padrões: estável (cerca de 75%), deterioração progressiva (cerca de 15%) e melhora (cerca de 10%). Mutações AIPL1 estão associadas à deterioração progressiva, enquanto mutações RPGRIP1 estão associadas a um curso estável. No futuro, espera-se a interrupção da progressão ou tratamento por visão artificial, terapia gênica e medicina regenerativa.
Duan W, Zhou T, Jiang H, Zhang M, Hu M, Zhang L. A novel nonsense variant (c.1499C>G) in CRB1 caused Leber congenital amaurosis-8 in a Chinese family and a literature review. BMC Med Genomics. 2022;15(1):197. doi:10.1186/s12920-022-01356-z.
Mordà D, Alibrandi S, Scimone C, Rinaldi C, Scalinci SZ, Abate G, et al. Decoding pediatric inherited retinal dystrophies: Bridging genetic complexity and clinical heterogeneity. Progress in retinal and eye research. 2025;109:101405. doi:10.1016/j.preteyeres.2025.101405. PMID:40998214.
Botto C, Rucli M, Tekinsoy MD, Pulman J, Sahel JA, Dalkara D. Early and late stage gene therapy interventions for inherited retinal degenerations. Progress in retinal and eye research. 2022;86:100975. doi:10.1016/j.preteyeres.2021.100975. PMID:34058340.
Gurnani B, et al. Clinical approach to pediatric nystagmus: a comprehensive diagnostic algorithm. Clin Ophthalmol. 2025;19:1617-1636.
Russell S, Bennett J, Wellman JA, Chung DC, Yu ZF, Tillman A, et al. Efficacy and safety of voretigene neparvovec (AAV2-hRPE65v2) in patients with RPE65-mediated inherited retinal dystrophy: a randomised, controlled, open-label, phase 3 trial. Lancet (London, England). 2017;390(10097):849-860. doi:10.1016/S0140-6736(17)31868-8. PMID:28712537; PMCID:PMC5726391.
Maguire AM, Russell S, Wellman JA, Chung DC, Yu ZF, Tillman A, et al. Efficacy, Safety, and Durability of Voretigene Neparvovec-rzyl in RPE65 Mutation-Associated Inherited Retinal Dystrophy: Results of Phase 1 and 3 Trials. Ophthalmology. 2019;126(9):1273-1285. doi:10.1016/j.ophtha.2019.06.017. PMID:31443789.
Fujinami K, Akiyama K, Tsunoda K, et al. Efficacy and Safety of Voretigene Neparvovec in RPE65-Retinopathy: Results of a Phase III Trial in Japan. Ophthalmol Sci. 2025;5:100876. doi:10.1016/j.xops.2025.100876. PMID:40910102; PMCID:PMC12405627.
Gange WS, Sisk RA, Besirli CG, Lee TC, Havunjian M, Schwartz H, et al. Perifoveal Chorioretinal Atrophy after Subretinal Voretigene Neparvovec-rzyl for RPE65-Mediated Leber Congenital Amaurosis. Ophthalmology. Retina. 2022;6(1):58-64. doi:10.1016/j.oret.2021.03.016. PMID:33838313; PMCID:PMC8497635.
Fischer MD, Simonelli F, Sahni J, Holz FG, Maier R, Fasser C, et al. Real-World Safety and Effectiveness of Voretigene Neparvovec: Results up to 2 Years from the Prospective, Registry-Based PERCEIVE Study. Biomolecules. 2024;14(1). doi:10.3390/biom14010122. PMID:38254722; PMCID:PMC10813228.
Copie o texto do artigo e cole no assistente de IA de sua preferência.
Artigo copiado para a área de transferência
Abra um assistente de IA abaixo e cole o texto copiado na conversa.