1869年由德国眼科医生特奥多尔·卡尔·古斯塔夫·冯·莱伯(Theodor Karl Gustav von Leber, 1840–1917)首次报道。需要注意的是,同一位莱伯于1871年报道的莱伯遗传性视神经病变(LHON)是一种线粒体疾病,发病年龄在20岁左右,与LCA完全不同。1957年,视网膜电图(ERG)波形消失被确认为LCA诊断的共同特征,疾病名称得以确立。
Higa N, et al. A novel RPE65 variant p.(Ala391Asp) in Leber congenital amaurosis: a case report and literature review in Japan. Front Med (Lausanne). 2024. Figure 1. PMCID: PMC11446184. License: CC BY.
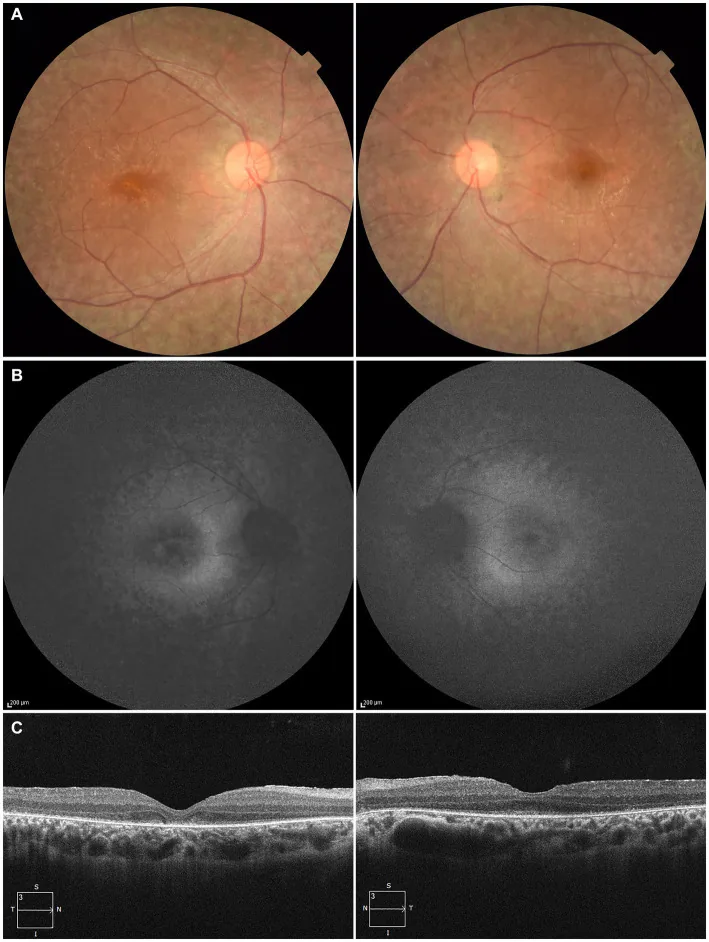
Duan W, Zhou T, Jiang H, Zhang M, Hu M, Zhang L. A novel nonsense variant (c.1499C>G) in CRB1 caused Leber congenital amaurosis-8 in a Chinese family and a literature review. BMC Med Genomics. 2022;15(1):197. doi:10.1186/s12920-022-01356-z.
Mordà D, Alibrandi S, Scimone C, Rinaldi C, Scalinci SZ, Abate G, et al. Decoding pediatric inherited retinal dystrophies: Bridging genetic complexity and clinical heterogeneity. Progress in retinal and eye research. 2025;109:101405. doi:10.1016/j.preteyeres.2025.101405. PMID:40998214.
Botto C, Rucli M, Tekinsoy MD, Pulman J, Sahel JA, Dalkara D. Early and late stage gene therapy interventions for inherited retinal degenerations. Progress in retinal and eye research. 2022;86:100975. doi:10.1016/j.preteyeres.2021.100975. PMID:34058340.
Gurnani B, et al. Clinical approach to pediatric nystagmus: a comprehensive diagnostic algorithm. Clin Ophthalmol. 2025;19:1617-1636.
Russell S, Bennett J, Wellman JA, Chung DC, Yu ZF, Tillman A, et al. Efficacy and safety of voretigene neparvovec (AAV2-hRPE65v2) in patients with RPE65-mediated inherited retinal dystrophy: a randomised, controlled, open-label, phase 3 trial. Lancet (London, England). 2017;390(10097):849-860. doi:10.1016/S0140-6736(17)31868-8. PMID:28712537; PMCID:PMC5726391.
Maguire AM, Russell S, Wellman JA, Chung DC, Yu ZF, Tillman A, et al. Efficacy, Safety, and Durability of Voretigene Neparvovec-rzyl in RPE65 Mutation-Associated Inherited Retinal Dystrophy: Results of Phase 1 and 3 Trials. Ophthalmology. 2019;126(9):1273-1285. doi:10.1016/j.ophtha.2019.06.017. PMID:31443789.
Fujinami K, Akiyama K, Tsunoda K, et al. Efficacy and Safety of Voretigene Neparvovec in RPE65-Retinopathy: Results of a Phase III Trial in Japan. Ophthalmol Sci. 2025;5:100876. doi:10.1016/j.xops.2025.100876. PMID:40910102; PMCID:PMC12405627.
Gange WS, Sisk RA, Besirli CG, Lee TC, Havunjian M, Schwartz H, et al. Perifoveal Chorioretinal Atrophy after Subretinal Voretigene Neparvovec-rzyl for RPE65-Mediated Leber Congenital Amaurosis. Ophthalmology. Retina. 2022;6(1):58-64. doi:10.1016/j.oret.2021.03.016. PMID:33838313; PMCID:PMC8497635.
Fischer MD, Simonelli F, Sahni J, Holz FG, Maier R, Fasser C, et al. Real-World Safety and Effectiveness of Voretigene Neparvovec: Results up to 2 Years from the Prospective, Registry-Based PERCEIVE Study. Biomolecules. 2024;14(1). doi:10.3390/biom14010122. PMID:38254722; PMCID:PMC10813228.