Leber konjenital amaurosis (LCA), doğumdan bebeklik dönemine kadar ciddi görme bozukluğuna neden olan konjenital retinal distrofilerin en şiddetli formudur. Çocukluk çağı görme bozukluğunun önemli bir nedenidir ve konjenital görme kaybının tipik bir temsilcisidir. Klinik görünüm çeşitlidir.
İlk kez 1869’da Alman göz doktoru Theodor Karl Gustav von Leber (1840–1917) tarafından tanımlanmıştır. Aynı Leber’in 1871’de tanımladığı Leber herediter optik nöropati (LHON), yaklaşık 20 yaşında başlayan mitokondriyal bir hastalıktır ve LCA’dan tamamen farklıdır. 1957’de elektroretinografide (ERG) dalga kaybının LCA tanısında ortak bir özellik olduğu doğrulanmış ve hastalık adı yerleşmiştir.
Tahmini doğum prevalansı 100.000 doğumda 2-3 kişidir (1/30.000 - 1/81.000)1). Bazı kaynaklar 1:80.000 - 1:200.000 arasında değişen oranlar bildirmektedir2). Tüm retinal distrofilerin yaklaşık %5’ini oluşturur ve körler okuluna devam eden görme engelli çocukların yaklaşık %20’si LCA’lıdır1). Şu ana kadar yaklaşık 27 LCA ilişkili gen tanımlanmıştır2) ve vakaların yaklaşık %70-80’inde sorumlu gen belirlenebilmektedir2). Kalıtım şekli çoğunlukla otozomal resesiftir, ancak otozomal dominant ve X’e bağlı kalıtım da bildirilmiştir.
QLeber konjenital amaurosis genellikle ne zaman fark edilir?
A
Genellikle ebeveynler, bebek yaklaşık 6 haftalıkken nistagmus veya sabit bakış eksikliği nedeniyle durumu fark eder3). Şiddetli görsel yanıt yokluğu (sabit bakış ve takibin tamamen olmaması) durumunda LCA’dan şüphelenilir ve elektroretinografi ile kesin tanı konur.
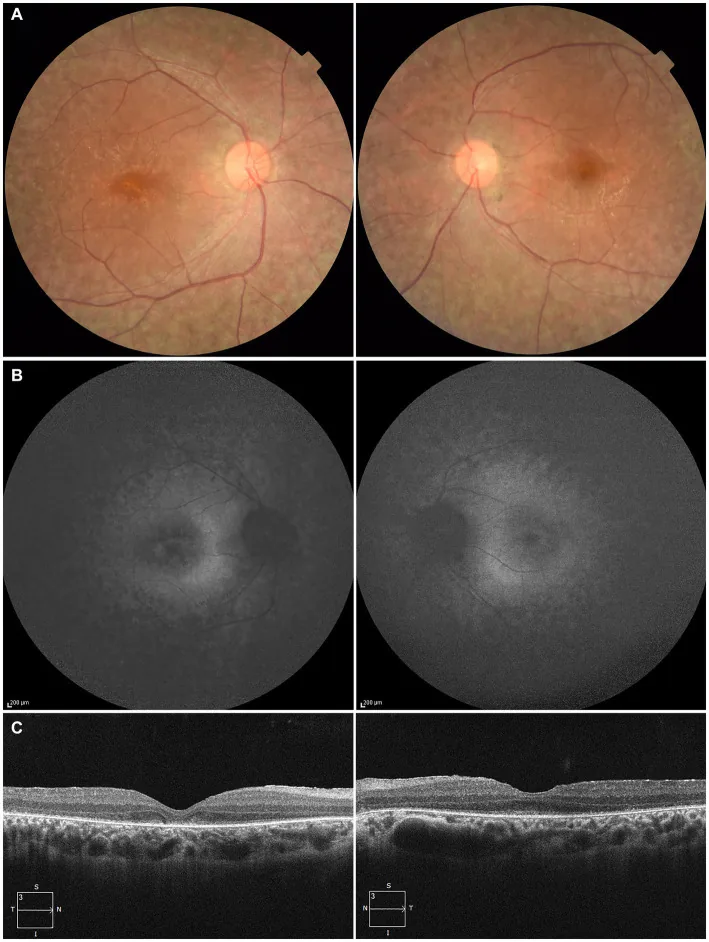
Higa N, et al. A novel RPE65 variant p.(Ala391Asp) in Leber congenital amaurosis: a case report and literature review in Japan. Front Med (Lausanne). 2024. Figure 1. PMCID: PMC11446184. License: CC BY.
27 yaşındaki bir hastada, (A) fundus fotoğrafında her iki gözde retina pigment epitelinde depigmentasyon ve retina damarlarında incelme, (B) fundus otofloresansında lezyon bölgelerinde hiperfloresans ve sinyal azalması, (C) OCT’de sağ gözde elipsoid zon (EZ) kaybı ve sol gözde foveada hafif kalıntı görülmektedir. Bu bulgular, metnin “2. Ana belirtiler ve klinik bulgular” bölümünde ele alınan retina pigment epitel dejenerasyonu ve retina damar incelmesi ile uyumludur.
Doğumdan veya erken postnatal dönemden itibaren ciddi görme bozukluğu mevcuttur.
Görme azlığı: Hastaların çoğunda görme keskinliği 0.1’in altındadır ve yaklaşık üçte birinde ışık hissi yoktur. Görme bozukluğu genellikle stabildir veya çok yavaş ilerler.
Fotofobi: Birçok hasta ışığa karşı aşırı duyarlılık gösterir.
Gece körlüğü: Karanlıkta görme işlevi daha da azalır.
Ebeveynler genellikle doğumdan 6 hafta sonra nistagmus veya sabit bakış eksikliği nedeniyle fark eder3).
Nistagmus: Doğumda veya hemen sonrasında ortaya çıkar. Sarkaç benzeri veya gezici tiptedir ve tüm bakış yönlerinde görülür. Erken başlangıçlı şiddetli retina distrofisinden (EOSRD) ayırt edilmesinde önemli bir noktadır; EOSRD’de nistagmus görülmez2).
Anormal pupilla reaksiyonu: Işık refleksi yavaşlamış veya kaybolmuştur. Buna “amaurotik pupilla” denir.
Okülo-dijital işaret: Gözleri dürtme, itme veya ovuşturma hareketleri; retinanın mekanik olarak uyarılması ve görsel duyum oluşturma girişimi olduğu düşünülmektedir. Başlıca sekeli, orbital yağ atrofisine bağlı enoftalmi (göz çökmesi)dir.
Refraksiyon kusurları: Şiddetli hipermetropi (>5 diyoptri) yaygındır ve erken görme bozukluğuna bağlı emetropizasyon bozukluğundan kaynaklandığı düşünülmektedir.
Bebeklik döneminde fundus sıklıkla normal görünür, ancak daha sonra çeşitli bulgular ortaya çıkar. Fundus bulguları normal fundustan tipik retinitis pigmentoza benzeri fundusa kadar çok çeşitlidir. İlerlemiş vakalarda optik disk soluklaşır, damarlar aşırı incelir, fundusun genel rengi koyulaşır ve makula halka refleksi kaybolur.
Optik disk solukluğu ve retina damarlarında incelme
Tuz-biber fundus: Küçük beyaz lekeler ve depigmentasyon ile retina pigment epitel dejenerasyonu
Kemik dikeni şeklinde pigment birikimi ve subretinal flekler (mermer görünümlü fundus)
Makula dejenerasyonu ve Coats hastalığı benzeri eksüdasyon
OCT’de retina dış tabakaları neredeyse tamamen kaybolmuştur ve ellipsoid zon (IS/OS bağlantısı) incelmesi-kaybı karakteristiktir. Genotipe göre bulgular farklılık gösterir:
CRB1 mutasyonu: Retina kalınlığında artış (paradoksal bulgu, kaba laminasyon) 1)
QGöz kırpma belirtisi (gözleri ovuşturma hareketi) ne amaçla yapılır?
A
LCA hastalarına özgü bir davranıştır; gözleri parmakla iterek veya bastırarak retinayı mekanik olarak uyarıp görsel algıyı tetiklemeye çalıştıkları düşünülmektedir. Uzun süreli tekrar, orbital yağ atrofisine ve enoftalmiye neden olur.
LCA, kalıtsal retinal dejenerasyon hastalıkları grubudur ve çoğunluğu otozomal resesif kalıtım gösterir2). Nadiren CRX, IMPDH1 ve OTX2 mutasyonları otozomal dominant kalıtıma neden olur2). X’e bağlı kalıtım da rapor edilmiştir.
Günümüzde LCA1’den LCA19’a kadar 19 tipe ek olarak sekiz ilişkili gen daha rapor edilmiştir2). Nedensel genler, fotoreseptör morfogenezi, fototransdüksiyon silyumu, görsel döngü (visual cycle) gibi retina gelişimi ve işleviyle ilgili çoklu yolaklarda rol oynar.
LCA ile ilişkili genler beş ana fonksiyonel ağa sınıflandırılır2):
Retinoid metabolizması ve çubuk görsel döngüsü (RPE65, LRAT, RDH12)
Işık uyarısı algılama ve görsel algı (GUCY2D, CNGA3)
Fotoreseptör bağlantı siliumu ve dış segment bakımı (CEP290, RPGRIP1, RPGR)
Dünya çapında en sık görülen mutant genler ve oranları aşağıdaki gibidir:
Gen
Oran
İlgili yolak
CEP290
Yaklaşık %15
Silium işlevi
GUCY2D
Yaklaşık %12
Işık sinyal iletimi (cGMP sentezi)
CRB1
Yaklaşık %10
Hücre polaritesinin korunması
RPE65
Yaklaşık %8
Retinoid metabolizması
Bir Japon kohortunda (34 aile) NGS analizi yaklaşık %56 saptama oranı göstermiş olup en sık mutasyona uğrayan genler CRB1, NMNAT1 ve RPGRIP1 olarak rapor edilmiştir1).
Neden olan genin belirlenmesi, tedavi uygunluğunun değerlendirilmesiyle doğrudan ilişkilidir. Özellikle RPE65 mutasyonunun doğrulanması, gen tedavisi (voretigen neparvovek) için uygunluk kararında zorunludur2).
AIPL1, fosfodiesteraz 6’nın (PDE6) özel bir şaperonu olarak işlev görür; PDE6, fototransdüksiyonda cGMP’nin parçalanmasından sorumludur. AIPL1 eksikliği, PDE6 protein miktarında dramatik bir azalmaya, cGMP metabolizmasının bozulmasına, fotoreseptör dejenerasyonuna ve erken körlüğe yol açar2). AIPL1 mutasyonları tüm LCA vakalarının yaklaşık %5-10’unu oluşturur2).
QBir sonraki çocuğa LCA geçme olasılığı nedir?
A
Otozomal resesif kalıtımda, her iki ebeveyn de taşıyıcıysa, bir sonraki çocuğun etkilenme olasılığı %25, taşıyıcı olma olasılığı %50 ve etkilenmemiş/taşıyıcı olmama olasılığı %25’tir. Nedensel gen biliniyorsa doğum öncesi tanı veya preimplantasyon genetik tanı da seçenekler arasındadır.
LCA tanısı klinik olarak konur ve elektroretinografi ile kesin tanı ve genetik test ile moleküler genetik doğrulama gereklidir2).
Konjenital belirgin görsel yanıt yetersizliği (fiksasyon ve takip eksikliği) durumunda LCA’dan şüphelenilir. Şiddetli görme bozukluğu ve yüksek hipermetropisi olan bebeklerde, moleküler genetik test ile LCA araştırması ilk seçenektir4).
Alt tipe göre değişir (RPE65 tipinde kaybolur, GUCY2D tipinde normaldir)
Genetik test (NGS vb.)
Kesin tanı ve alt tip belirleme için gereklidir
Elektroretinografi: Çubuk ve koni yanıtlarının her ikisi de kaydedilemez veya belirgin şekilde azalmıştır. Normal elektroretinografi LCA tanısını dışlar.
OCT: Retinanın dış tabakaları ve ellipsoid zon neredeyse tamamen kaybolmuştur. CRB1 mutasyonlarında paradoksal retina kalınlaşması (kaba laminasyon) görülür1). El tipi OCT, uyanık bebeklerde veya anestezi altındaki küçük çocuklarda muayene için faydalıdır4).
FAF: Bulgular alt tipe göre değişir. GUCY2D mutasyonlarında otofloresans normal kalırken, RPE65 mutasyonlarında kaybolur2).
Genetik test: Yeni nesil dizileme (NGS), DNA mikrodizisi, bağlantı analizi vb. kullanılır. Genel tespit oranı yaklaşık %70-80’dir2). 2023’ten itibaren, 82 IRD nedensel genini içeren bir panel testi (PrismGuide IRD paneli) sigorta kapsamına alınmış ve RPE65 ile ilişkili IRD şüphesi olan genç bireylere uygulanmaktadır.
LCA’nın çoğu tipi için etkili bir tedavi mevcut değildir. Mevcut yönetim aşağıdaki gibidir:
Refraksiyon kusurlarının düzeltilmesi: Yüksek hipermetropi vb. için uygun refraktif düzeltme yapılır. Güçlü refraksiyon kusurları olabilir, bu nedenle gözlük reçetesi ve ortoptik eğitim önerilir.
Ortoptik eğitim: Kalan görsel işlevden maksimum düzeyde yararlanmak için görsel rehabilitasyon yapılır.
Görme yedek eğitimi: Görme işlevi ciddi şekilde bozulduğu için braille öğretimi, beyaz bastonla yürüme eğitimi ve büyüteçli okuma cihazlarının kullanımı da düşünülür.
Az görme bakımı: Az görme yardımcılarının kullanımı, eğitim ve iş fırsatlarına en uygun erişim desteklenir.
Genetik danışmanlık: Aile ve hastaya önerilir. Taşıyıcı testi, doğum öncesi tanı ve preimplantasyon genetik tanı mümkün olabilir.
Fotofobi yönetimi: Işık koruyucu gözlük kullanımı ve ışığa maruziyetin azaltılması önerilir.
Düzenli takip: Elektroretinogram dahil oftalmolojik takip ve gerektiğinde az görme kliniğine yönlendirme yapılır.
2017 yılında ABD Gıda ve İlaç Dairesi (FDA), biallelik RPE65 mutasyonlarıyla ilişkili LCA2 tedavisi için voretigen neparvovek-rzyl (ticari adı Luxturna)‘yı onayladı. Bu, oftalmoloji alanında FDA onaylı ilk gen tedavisi ürünüdür. 2023 yılında Japonya’da da onaylanmıştır (ürün adı: Luxturna® enjeksiyonu).
Normal RPE65 geninin bir kopyası, rekombinant adeno-ilişkili virüs (rAAV2) vektörü kullanılarak subretinal enjeksiyon yoluyla retina pigment epiteline aktarılır. İşlem vitrektomi ameliyathanesinde gerçekleştirilir.
Tam alan uyarı eşiğinde (FST) de anlamlı iyileşme gözlendi
Faz I/III uzun dönem sonuçları (Maguire 2019)6):
Tedaviden 6-12 ay sonra zirveye ulaşan retina hassasiyeti, görme keskinliği ve fonksiyonel kazanımların zamanla kademeli olarak azalma eğiliminde olduğu gösterildi
Retina hassasiyetindeki artışın gece körlüğü ve görme alanında iyileşme sağlaması beklenir
Yurt içi Faz III çalışması (A11301 çalışması) (Fujinami 2025)7):
Katılımcılar: RPE65 ile ilişkili IRD’li 4 Japon hasta
FST’de (tam alan uyarı eşiği) anlamlı hassasiyet artışı (10 kattan fazla hassasiyet artışı anlamlı olarak tanımlandı)
Uygulamadan bir yıl sonra görme alanında genişleme doğrulandı
Başlıca yan etkiler: Göz ağrısı dahil göz bozuklukları (uygulama tekniği ile ilişkili olduğu düşünülmektedir)
Uygulama protokolü:
İkinci göze uygulama, ilk gözden en az 6 gün sonra yapılır
İmmünsüpresyon: Uygulamadan 3 gün önce steroid başlanır ve uygulamadan sonra 14 gün devam edilir
Bununla birlikte, RPE65 mutasyonları tüm LCA hastalarının yalnızca yaklaşık %8’ini oluşturur. Diğer mutasyon tipleri için şu anda kanıtlanmış bir tedavi yoktur.
QGen tedavisi tüm LCA hastaları için kullanılabilir mi?
A
Şu anda onaylanmış gen tedavisi (voretigen neparvovek / Luxturna®) yalnızca iki alelik RPE65 mutasyonuna bağlı LCA2 için endikedir. RPE65 mutasyonları tüm LCA’ların yaklaşık %8’ini oluşturur ve hastaların büyük çoğunluğu için uygun değildir. Diğer genotipler için tedaviler araştırma aşamasındadır.
QJaponya'da gen tedavisi almak için ne yapılmalıdır?
A
İki alelik RPE65 mutasyonunun doğrulanması ve yeterli canlı retina hücresinin bulunması şarttır. Genetik panel testi (PrismGuide IRD paneli) ile genetik tanı ilk adımdır. Kalıtsal retina hastalıkları konusunda deneyimli uzman merkezlere yönlendirme önerilir.
6. Patofizyoloji ve ayrıntılı hastalık mekanizması
LCA’nın patofizyolojisi, görme döngüsünün (Visual Cycle) bozulmasıyla ilişkilidir ve bu da gözün ışık bilgisini iletememesine yol açar.
Görme döngüsü, retina pigment epiteli (RPE) ile nörosensoriyel retina arasında gerçekleşen bir dizi enzimatik reaksiyondur; diyetle alınan A vitaminini metabolize ederek 11-cis retinal üretir ve fotopigment oluşturur. 11-cis retinal olmadan, fototransdüksiyon kaskadı başlamaz ve görsel sinir sinyali görme korteksine iletilmez. Bu reaksiyon zincirinde yer alan proteinleri kodlayan genlerden herhangi birindeki mutasyon, görme döngüsünü bloke ederek LCA semptomlarına neden olur.
Histopatolojik olarak, dış retina ve fotoreseptörlerin tutulumu gösterilmiştir ve LCA’nın bir gelişimsel bozukluktan ziyade dejeneratif bir süreç olduğu düşünülmektedir.
GUCY2D (LCA1): Retinaya özgü guanilat siklazı (GC-E) kodlar. cGMP sentezini katalize eder ve ışık sinyal iletiminin anahtarıdır. 140’tan fazla hastalıkla ilişkili mutasyon tanımlanmıştır ve %88’i otozomal resesif LCA’nın nedenidir. 838. amino asit mutasyon sıcak noktası olarak bilinir 2).
RPE65 (LCA2): Karotenoid kesen oksijenaz süperailesine aittir ve all-trans-retinil esterden O-alkil ester kesimi ve retinil kısmının izomerizasyonunu (all-trans-retinol → 11-cis-retinol) katalize eden çift işlevli bir enzimdir 2). Hem çubuk hem de koni hücre işlevi için gereklidir ve son zamanlarda luteinden mezo-zeaksantine izomerizasyonda da rol oynayabileceği öne sürülmüştür 2). Gen tedavisi için onaylanmış tek endikasyon.
CRB1 (LCA8): Meyve sineğindeki crumbs proteini ile homologdur, fotoreseptör iç segmenti ve Müller hücrelerinde eksprese edilir. Hücre polaritesinin korunması için önemlidir ve kromozom 1q31.3’te yer alır 1).
CEP290 (LCA10): Fotoreseptör silya işlevinde rol oynar. LCA ile ilişkili genler arasında en yüksek mutasyon sıklığına sahiptir (yaklaşık %15).
LCA5: Lebercilin proteinini kodlar ve silya işlevi ile silya içi protein taşınmasında rol oynar 2).
AIPL1: PDE6 (fosfodiesteraz 6) için özel bir şaperon olarak işlev görür. AIPL1 eksikliği PDE6’nın kararsızlaşmasına → cGMP metabolizmasının bozulmasına → kanal anormalliğine → fotoreseptör dejenerasyonuna yol açar 2).
7. Güncel araştırmalar ve geleceğe bakış (araştırma aşamasındaki raporlar)
Voretigen neparvovek’in uzun dönem takip değerlendirmesinde (NCT00481546, NCT00643747), tedaviden 6-12 ay sonra erken bir zirve görüldükten sonra retina hassasiyeti, görme keskinliği ve fonksiyonel kazanç dahil klinik faydaların giderek azaldığı bildirilmiştir 6). PERCEIVE çalışması (prospektif kayıt çalışması), gerçek klinikte 2 yıllık güvenlik ve etkinlik verilerini raporlamış ve vakaların en fazla %50’sinde gen tedavisi ilişkili üveit (GTAU) gözlenmiştir 9).
Editas Medicine tarafından geliştirilmiştir. AAV5 vektörü, S. aureus kaynaklı Cas9 ve iki kılavuz RNA taşır ve CEP290’ın intron 26’sındaki derin intron mutasyonunu (c.2991+1655A>G) hedefler 3). İlk insan çalışmasında güvenlik doğrulanmış ve nispeten yüksek dozlarda bile iyi tolere edildiği gösterilmiştir 3).
Optogenetik ile Görme Fonksiyonunun Geri Kazanılması
Gen bağımsız bir yaklaşım olarak, kalan iç retina nöronlarında ışığa duyarlı kanalrodopsinlerin ifade edilmesi yöntemi araştırılmaktadır 3). Tüm LCA tiplerine (genotipten bağımsız olarak) potansiyel olarak uygulanabilir ve erken klinik denemeler devam etmektedir.
GUCY2D ve AIPL1 mutasyonlarına yönelik gen tedavisi hayvan modellerinde devam etmekte olup, çubuk ve koni fotoreseptörlerinin kurtarılmasında umut verici sonuçlar göstermiştir.
LCA’nın seyri üç paterne ayrılır: stabil (yaklaşık %75), ilerleyici kötüleşme (yaklaşık %15) ve iyileşme (yaklaşık %10). AIPL1 mutasyonu ilerleyici kötüleşme ile, RPGRIP1 mutasyonu ise stabil seyir ile ilişkilidir. Gelecekte yapay görme, gen tedavisi ve rejeneratif tıp ile ilerlemenin durdurulması veya tedavi beklenmektedir.
Duan W, Zhou T, Jiang H, Zhang M, Hu M, Zhang L. A novel nonsense variant (c.1499C>G) in CRB1 caused Leber congenital amaurosis-8 in a Chinese family and a literature review. BMC Med Genomics. 2022;15(1):197. doi:10.1186/s12920-022-01356-z.
Mordà D, Alibrandi S, Scimone C, Rinaldi C, Scalinci SZ, Abate G, et al. Decoding pediatric inherited retinal dystrophies: Bridging genetic complexity and clinical heterogeneity. Progress in retinal and eye research. 2025;109:101405. doi:10.1016/j.preteyeres.2025.101405. PMID:40998214.
Botto C, Rucli M, Tekinsoy MD, Pulman J, Sahel JA, Dalkara D. Early and late stage gene therapy interventions for inherited retinal degenerations. Progress in retinal and eye research. 2022;86:100975. doi:10.1016/j.preteyeres.2021.100975. PMID:34058340.
Gurnani B, et al. Clinical approach to pediatric nystagmus: a comprehensive diagnostic algorithm. Clin Ophthalmol. 2025;19:1617-1636.
Russell S, Bennett J, Wellman JA, Chung DC, Yu ZF, Tillman A, et al. Efficacy and safety of voretigene neparvovec (AAV2-hRPE65v2) in patients with RPE65-mediated inherited retinal dystrophy: a randomised, controlled, open-label, phase 3 trial. Lancet (London, England). 2017;390(10097):849-860. doi:10.1016/S0140-6736(17)31868-8. PMID:28712537; PMCID:PMC5726391.
Maguire AM, Russell S, Wellman JA, Chung DC, Yu ZF, Tillman A, et al. Efficacy, Safety, and Durability of Voretigene Neparvovec-rzyl in RPE65 Mutation-Associated Inherited Retinal Dystrophy: Results of Phase 1 and 3 Trials. Ophthalmology. 2019;126(9):1273-1285. doi:10.1016/j.ophtha.2019.06.017. PMID:31443789.
Fujinami K, Akiyama K, Tsunoda K, et al. Efficacy and Safety of Voretigene Neparvovec in RPE65-Retinopathy: Results of a Phase III Trial in Japan. Ophthalmol Sci. 2025;5:100876. doi:10.1016/j.xops.2025.100876. PMID:40910102; PMCID:PMC12405627.
Gange WS, Sisk RA, Besirli CG, Lee TC, Havunjian M, Schwartz H, et al. Perifoveal Chorioretinal Atrophy after Subretinal Voretigene Neparvovec-rzyl for RPE65-Mediated Leber Congenital Amaurosis. Ophthalmology. Retina. 2022;6(1):58-64. doi:10.1016/j.oret.2021.03.016. PMID:33838313; PMCID:PMC8497635.
Fischer MD, Simonelli F, Sahni J, Holz FG, Maier R, Fasser C, et al. Real-World Safety and Effectiveness of Voretigene Neparvovec: Results up to 2 Years from the Prospective, Registry-Based PERCEIVE Study. Biomolecules. 2024;14(1). doi:10.3390/biom14010122. PMID:38254722; PMCID:PMC10813228.
Makale metnini kopyalayıp tercih ettiğiniz yapay zeka asistanına yapıştırabilirsiniz.
Makale panoya kopyalandı
Aşağıdaki yapay zeka asistanlarından birini açın ve kopyalanan metni sohbet kutusuna yapıştırın.