L’amaurosi congenita di Leber (LCA) è la forma più grave di distrofia retinica congenita, causando un grave deficit visivo dalla nascita o nell’infanzia. È una delle principali cause di deficit visivo infantile ed è nota come causa rappresentativa di cecità congenita. Il quadro clinico è vario.
Fu descritta per la prima volta nel 1869 dall’oftalmologo tedesco Theodor Karl Gustav von Leber (1840–1917). La neuropatia ottica ereditaria di Leber (LHON), descritta dallo stesso Leber nel 1871, è una malattia mitocondriale che insorge intorno ai 20 anni ed è completamente diversa dalla LCA. Nel 1957, l’assenza di onde all’elettroretinogramma (ERG) fu confermata come caratteristica comune della diagnosi di LCA, stabilendo il nome della malattia.
La prevalenza stimata alla nascita è di 2-3 per 100.000 nati (1/30.000 - 1/81.000)1). Alcune fonti riportano valori da 1:80.000 a 1:200.000, indicando variabilità2). Rappresenta circa il 5% di tutte le distrofie retiniche e circa il 20% dei bambini ipovedenti che frequentano scuole per ciechi ha LCA1). Attualmente sono stati identificati circa 27 geni associati alla LCA2) e in circa il 70-80% dei casi viene identificato il gene causale2). La modalità di trasmissione è principalmente autosomica recessiva, ma sono stati riportati casi di trasmissione dominante e legata all’X.
QQuando viene solitamente riconosciuta l'amaurosi congenita di Leber?
A
Di solito i genitori notano nistagmo o assenza di fissazione intorno alle 6 settimane di età3). In caso di grave deficit di reazione visiva (nessuna fissazione o inseguimento), si sospetta LCA, confermata dall’elettroretinogramma.
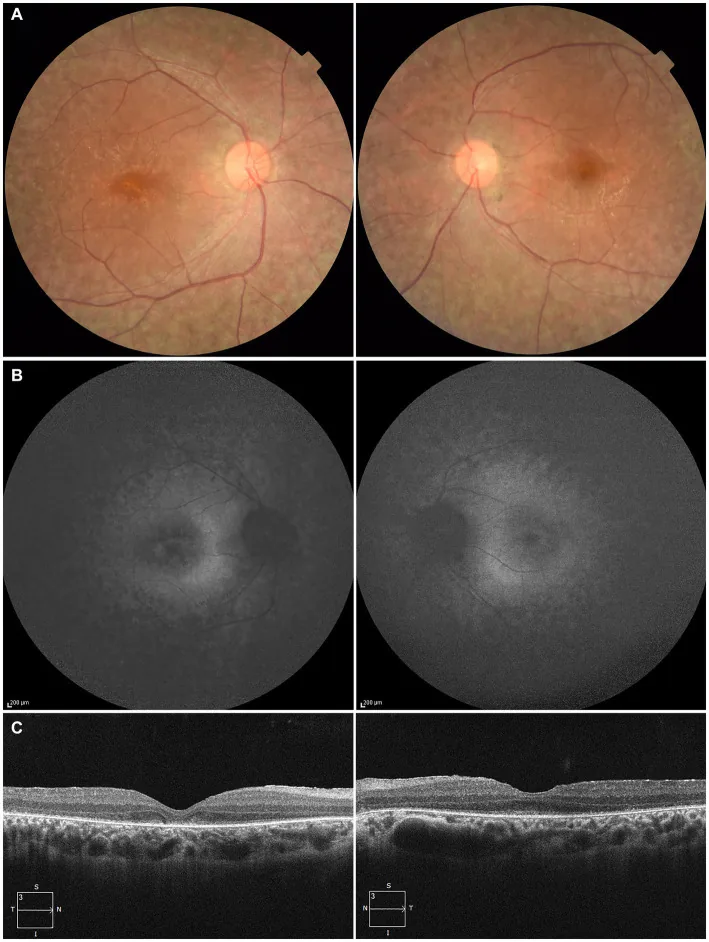
Higa N, et al. A novel RPE65 variant p.(Ala391Asp) in Leber congenital amaurosis: a case report and literature review in Japan. Front Med (Lausanne). 2024. Figure 1. PMCID: PMC11446184. License: CC BY.
In un paziente di 27 anni, (A) le fotografie del fondo oculare di entrambi gli occhi mostrano depigmentazione dell’epitelio pigmentato retinico e restringimento dei vasi retinici, (B) l’autofluorescenza del fondo mostra iperfluorescenza e riduzione del segnale nelle aree lesionali, (C) l’OCT mostra la scomparsa della zona ellissoidale (EZ) nell’occhio destro e un lieve residuo nella fovea dell’occhio sinistro. Ciò corrisponde alla degenerazione dell’epitelio pigmentato retinico e al restringimento dei vasi retinici trattati nella sezione «2. Principali sintomi e segni clinici».
Un grave deficit visivo è presente dalla nascita o subito dopo.
Riduzione dell’acuità visiva: La maggior parte dei pazienti ha un’acuità visiva inferiore a 0,1 e circa un terzo non ha percezione luminosa. Il deficit visivo è generalmente stabile o progredisce molto lentamente.
Fotofobia: Molti pazienti mostrano ipersensibilità alla luce.
Eméralopia: La funzione visiva al buio è ulteriormente ridotta.
I genitori spesso notano nistagmo o mancanza di fissazione intorno alle 6 settimane di età 3).
Nistagmo: Compare alla nascita o subito dopo. È pendolare o errante, presente in tutte le posizioni dello sguardo. È un importante punto di differenziazione dalla distrofia retinica grave a esordio precoce (EOSRD), che non si accompagna a nistagmo2).
Anomalia della reazione pupillare: Il riflesso fotomotore è lento o assente. Si parla di «pupille amaurotiche».
Segno oculo-digitale: Il paziente si picchia, preme o strofina gli occhi, considerato un tentativo di stimolare meccanicamente la retina per evocare visione. La principale conseguenza è l’enoftalmo da atrofia del grasso orbitario.
Errore refrattivo: L’ipermetropia elevata (>5 diottrie) è comune, probabilmente dovuta a un’alterazione dell’emmetropizzazione causata dal deficit visivo precoce.
Nell’infanzia il fondo oculare può spesso apparire normale, ma successivamente compaiono reperti vari. I reperti del fondo vanno da un fondo normale a un aspetto tipico di retinite pigmentosa. Nei casi avanzati, la papilla ottica diventa pallida, i vasi sono molto ristretti, il tono complessivo del fondo si scurisce e il riflesso anulare maculare scompare.
Pallore della papilla ottica e restringimento dei vasi retinici
Fondo a sale e pepe: piccole macchie bianche e depigmentazione dell’epitelio pigmentato retinico
Pigmentazione a spicole ossee e drusen sottoretinici (fondo marmorizzato)
All’OCT gli strati retinici esterni sono quasi assenti, con assottigliamento o scomparsa della zona ellissoidale (giunzione IS/OS). I reperti variano in base al genotipo:
Mutazione CRB1: ispessimento paradossale della retina (lamination grossolana) 1)
Mutazione RPE65: scomparsa dell’autofluorescenza del fondo (FAF) 2)
QA cosa serve il segno oculo-digitale (sfregamento degli occhi)?
A
È un comportamento caratteristico dei pazienti con LCA: si strofinano o premono gli occhi con le dita per stimolare meccanicamente la retina e cercare di evocare una sensazione visiva. La ripetizione a lungo termine porta all’atrofia del grasso orbitario e può causare enoftalmo.
La LCA è un gruppo di malattie degenerative ereditarie della retina, la maggior parte delle quali segue una trasmissione autosomica recessiva2). Raramente, mutazioni in CRX, IMPDH1 o OTX2 possono mostrare una trasmissione autosomica dominante2). Sono stati riportati anche casi di trasmissione legata all’X.
Attualmente sono descritti 19 tipi di LCA (LCA1–LCA19) e altri 8 geni associati2). I geni causali sono coinvolti in diverse vie dello sviluppo e della funzione retinica, tra cui la morfogenesi dei fotorecettori, i cigli della fototrasduzione e il ciclo visivo.
I geni associati alla LCA sono classificati in cinque principali reti funzionali2):
Metabolismo dei retinoidi e ciclo visivo dei bastoncelli (RPE65, LRAT, RDH12)
Mantenimento dell’omeostasi retinica e dei fotorecettori (AIPL1, SPATA7, TULP1, USP45, CRB1, LCA5)
Sviluppo e morfogenesi retinica (RD3, CEP290)
Rilevamento dello stimolo luminoso e percezione visiva (GUCY2D, CNGA3)
Ciglia connettive dei fotorecettori e mantenimento del segmento esterno (CEP290, RPGRIP1, RPGR)
I geni mutati più frequenti a livello mondiale e le loro proporzioni sono i seguenti:
Gene
Proporzione
Via coinvolta
CEP290
Circa il 15%
Funzione ciliare
GUCY2D
Circa il 12%
Trasduzione del segnale luminoso (sintesi di cGMP)
CRB1
Circa il 10%
Mantenimento della polarità cellulare
RPE65
circa l’8%
Metabolismo dei retinoidi
In una coorte giapponese (34 famiglie), l’analisi NGS ha mostrato un tasso di rilevamento di circa il 56%, con i geni mutati più frequenti CRB1, NMNAT1 e RPGRIP11).
L’identificazione del gene causale è direttamente collegata alla determinazione dell’idoneità al trattamento. In particolare, la conferma di una mutazione RPE65 è essenziale per valutare l’idoneità alla terapia genica (voretigene neparvovec)2).
AIPL1 funge da chaperone specifico per la fosfodiesterasi 6 (PDE6), che degrada il cGMP nella trasduzione del segnale luminoso. La carenza di AIPL1 porta a una drastica riduzione della proteina PDE6, con conseguente alterazione del metabolismo del cGMP, degenerazione dei fotorecettori e cecità precoce2). Le mutazioni di AIPL1 rappresentano circa il 5-10% di tutte le LCA2).
QQual è la probabilità che il prossimo bambino erediti la LCA?
A
In caso di ereditarietà autosomica recessiva, se entrambi i genitori sono portatori, la probabilità che il prossimo bambino sia affetto è del 25%, che sia portatore del 50% e che non sia né affetto né portatore del 25%. Se il gene causale è noto, sono possibili la diagnosi prenatale o la diagnosi preimpianto.
La diagnosi di LCA è clinica e richiede la conferma mediante elettroretinografia e la conferma molecolare mediante test genetico2).
La LCA è sospettata in caso di grave riduzione della risposta visiva congenita (assenza di fissazione e inseguimento). Nei neonati con grave deficit visivo e ipermetropia elevata, la ricerca di LCA mediante test genetico molecolare è la prima scelta4).
I principali esami diagnostici sono elencati di seguito.
Variabile a seconda del sottotipo (nel tipo RPE65 scompare, nel tipo GUCY2D è normale).
Test genetico (NGS, ecc.)
Necessario per la diagnosi definitiva e l’identificazione del sottotipo.
Elettroretinogramma: Le risposte dei bastoncelli e dei coni sono assenti o marcatamente ridotte. Un ERG normale esclude la diagnosi di LCA.
OCT: Gli strati esterni della retina e la zona ellissoidale sono quasi scomparsi. Nelle mutazioni CRB1 si osserva un paradosso ispessimento retinico (lamellazione grossolana)1). L’OCT portatile è utile per l’esame di neonati svegli o bambini piccoli in anestesia4).
FAF: I reperti variano a seconda del sottotipo. Nelle mutazioni GUCY2D l’autofluorescenza è conservata normale, mentre nelle mutazioni RPE65 scompare2).
Test genetico: Vengono utilizzati il sequenziamento di nuova generazione (NGS), i microarray di DNA, l’analisi di linkage, ecc. Il tasso di rilevamento complessivo è di circa il 70-80%2). Dal 2023, un test panel per 82 geni patogeni della IRD (pannello PrismGuide IRD) è coperto dall’assicurazione sanitaria e si applica a giovani pazienti con sospetta IRD correlata a RPE65.
Per la maggior parte delle forme di LCA non esiste una terapia curativa consolidata. La gestione attuale è la seguente:
Correzione dei vizi di refrazione : Correzione appropriata per ipermetropia elevata, ecc. Può essere presente un forte vizio refrattivo, quindi prescrivere occhiali e incoraggiare l’ortottica.
Ortottica : Riabilitazione visiva per massimizzare l’uso della funzione visiva residua.
Addestramento all’uso di ausili visivi : A causa della significativa compromissione della funzione visiva, considerare l’insegnamento del Braille, l’addestramento al bastone bianco, l’uso di ingranditori elettronici, ecc.
Nel 2017, la FDA statunitense ha approvato voretigene neparvovec-rzyl (nome commerciale Luxturna) per il trattamento della LCA2 associata a mutazioni bialleliche di RPE65. È il primo prodotto di terapia genica approvato dalla FDA in oftalmologia. Nel 2023 è stato approvato anche in Giappone (nome del prodotto: Luxturna®).
Un vettore virale adeno-associato ricombinante (rAAV2) viene utilizzato per introdurre una copia normale del gene RPE65 nell’epitelio pigmentato retinico mediante iniezione sottoretinica. La procedura viene eseguita in sala operatoria per vitrectomia.
Studio di fase III 301 (Russell 2017)5):
Soggetti: 31 pazienti con IRD correlata a RPE65
Endpoint primario: MLMT (test di mobilità a più luminanze)
Miglioramento significativo osservato anche nel test di soglia di stimolazione a campo pieno (FST)
Risultati a lungo termine di fase I/III (Maguire 2019)6):
La sensibilità retinica, l’acuità visiva e i guadagni funzionali, che raggiungono il picco 6-12 mesi dopo il trattamento, hanno mostrato una tendenza a diminuire progressivamente in seguito.
L’aumento della sensibilità retinica può migliorare la cecità notturna e il campo visivo.
Studio nazionale di fase III (A11301) (Fujinami 2025)7):
Soggetti: 4 pazienti giapponesi con IRD correlata a RPE65
Aumento significativo della sensibilità al FST (definito come aumento della sensibilità di oltre 10 volte)
Espansione del campo visivo confermata a 1 anno dalla somministrazione.
Principali eventi avversi: disturbi oculari inclusi dolore oculare (presumibilmente correlati alla procedura di somministrazione)
Protocollo di somministrazione:
Il secondo occhio viene trattato almeno 6 giorni dopo il primo
Immunosoppressione: iniziare gli steroidi 3 giorni prima della somministrazione, continuare per 14 giorni dopo
Tuttavia, le mutazioni RPE65 rappresentano solo circa l’8% di tutti i pazienti con LCA. Per altri tipi di mutazione, attualmente non esiste una terapia con efficacia dimostrata.
QLa terapia genica è utilizzabile per tutti i pazienti con LCA?
A
Attualmente, la terapia genica approvata (voretigene neparvovec / Luxturna®) è indicata solo per LCA2 causata da mutazioni bialleliche di RPE65. Le mutazioni RPE65 rappresentano circa l’8% di tutte le LCA e la maggior parte dei pazienti non ha indicazione. I trattamenti per altri genotipi sono in fase di ricerca.
QCome ricevere la terapia genica in Giappone?
A
Le condizioni sono la conferma di una mutazione biallelica di RPE65 e la presenza di sufficienti cellule retiniche vitali. La diagnosi genetica tramite pannello genico (PrismGuide IRD panel) è il primo passo. Si raccomanda il rinvio a un centro specializzato con esperienza nella gestione delle malattie retiniche ereditarie.
La fisiopatologia della LCA è legata all’interruzione del ciclo visivo (Visual Cycle), che impedisce all’occhio di trasmettere le informazioni luminose.
Il ciclo visivo è una serie di reazioni enzimatiche tra l’epitelio pigmentato retinico (RPE) e la retina neurosensoriale, che metabolizza la vitamina A alimentare per produrre 11-cis-retinale e generare pigmenti fotorecettori. Senza 11-cis-retinale, la cascata di fototrasduzione non può iniziare e i segnali nervosi visivi non vengono trasmessi alla corteccia visiva. Una mutazione in uno qualsiasi dei geni che codificano per le proteine coinvolte in questa serie di reazioni può bloccare il ciclo visivo e causare i sintomi della LCA.
Istopatologicamente, è stato dimostrato il coinvolgimento della retina esterna e dei fotorecettori, suggerendo che la LCA sia un processo degenerativo piuttosto che una displasia.
GUCY2D (LCA1) : codifica per la guanilato ciclasi specifica della retina (GC-E). Catalizza la sintesi di cGMP, chiave della fototrasduzione. Oltre 140 mutazioni patogenetiche identificate, l’88% causa di LCA autosomica recessiva. L’aminoacido 838 è noto come hot spot mutazionale 2).
RPE65 (LCA2) : appartiene alla superfamiglia delle carotenoidi-ossigenasi. È un enzima bifunzionale che catalizza il taglio dell’estere O-alchilico dall’all-trans-retinil estere e l’isomerizzazione della porzione retinilica (all-trans-retinolo → 11-cis-retinolo) 2). Essenziale per la funzione sia dei bastoncelli che dei coni. Studi recenti suggeriscono un possibile coinvolgimento nell’isomerizzazione della luteina in meso-zeaxantina 2). Unica indicazione approvata per la terapia genica.
CRB1 (LCA8) : omologo della proteina crumbs di Drosophila, espresso nel segmento interno dei fotorecettori e nelle cellule di Müller. Importante per il mantenimento della polarità cellulare, localizzato sul cromosoma 1q31.3 1).
CEP290 (LCA10) : coinvolto nella funzione ciliare dei fotorecettori. Il gene più frequentemente mutato tra quelli associati a LCA (circa 15%).
NMNAT1 (LCA9) : codifica per un enzima chiave della biosintesi del NAD (nicotinammide adenina dinucleotide) 2).
LCA5 : codifica per la lebercilina, coinvolta nella funzione ciliare e nel trasporto intraciliare delle proteine 2).
AIPL1 : funge da chaperone specifico per PDE6 (fosfodiesterasi 6). La carenza di AIPL1 destabilizza PDE6 → alterazione del metabolismo del cGMP → anomalia dei canali → degenerazione dei fotorecettori 2).
7. Ricerche recenti e prospettive future (rapporti in fase di ricerca)
La valutazione di follow-up a lungo termine del voretigene neparvovec (NCT00481546, NCT00643747) riporta un picco iniziale a 6-12 mesi dopo il trattamento, seguito da una progressiva diminuzione dei benefici clinici, tra cui sensibilità retinica, acuità visiva e guadagni funzionali 6). Lo studio PERCEIVE (studio prospettico di registro) ha riportato dati di sicurezza ed efficacia a 2 anni nella pratica clinica reale, con uveite associata a terapia genica (GTAU) osservata in fino al 50% dei casi 9).
Sviluppato da Editas Medicine. Il vettore AAV5 trasporta Cas9 di S. aureus e due RNA guida, mirando alla mutazione intronica profonda (c.2991+1655A>G) nell’introne 26 di CEP2903). Lo studio first-in-human ha confermato la sicurezza e una buona tollerabilità anche a dosi relativamente elevate3).
Recupero della funzione visiva tramite optogenetica
Come approccio gene-indipendente, viene studiato un metodo per esprimere la channelrhodopsina fotosensibile nei neuroni retinici interni residui3). Potrebbe essere applicabile a tutti i tipi di LCA (indipendentemente dal genotipo) e sono in corso studi clinici iniziali.
La terapia genica per le mutazioni GUCY2D e AIPL1 è in corso in modelli animali, mostrando risultati promettenti nel salvataggio dei fotorecettori a bastoncelli e coni.
Il decorso della LCA è classificato in tre modelli: stabile (circa 75%), peggioramento progressivo (circa 15%) e miglioramento (circa 10%). Le mutazioni AIPL1 sono associate a peggioramento progressivo, mentre le mutazioni RPGRIP1 sono correlate a un decorso stabile. In futuro si spera di arrestare la progressione o trattare con visione artificiale, terapia genica o medicina rigenerativa.
Duan W, Zhou T, Jiang H, Zhang M, Hu M, Zhang L. A novel nonsense variant (c.1499C>G) in CRB1 caused Leber congenital amaurosis-8 in a Chinese family and a literature review. BMC Med Genomics. 2022;15(1):197. doi:10.1186/s12920-022-01356-z.
Mordà D, Alibrandi S, Scimone C, Rinaldi C, Scalinci SZ, Abate G, et al. Decoding pediatric inherited retinal dystrophies: Bridging genetic complexity and clinical heterogeneity. Progress in retinal and eye research. 2025;109:101405. doi:10.1016/j.preteyeres.2025.101405. PMID:40998214.
Botto C, Rucli M, Tekinsoy MD, Pulman J, Sahel JA, Dalkara D. Early and late stage gene therapy interventions for inherited retinal degenerations. Progress in retinal and eye research. 2022;86:100975. doi:10.1016/j.preteyeres.2021.100975. PMID:34058340.
Gurnani B, et al. Clinical approach to pediatric nystagmus: a comprehensive diagnostic algorithm. Clin Ophthalmol. 2025;19:1617-1636.
Russell S, Bennett J, Wellman JA, Chung DC, Yu ZF, Tillman A, et al. Efficacy and safety of voretigene neparvovec (AAV2-hRPE65v2) in patients with RPE65-mediated inherited retinal dystrophy: a randomised, controlled, open-label, phase 3 trial. Lancet (London, England). 2017;390(10097):849-860. doi:10.1016/S0140-6736(17)31868-8. PMID:28712537; PMCID:PMC5726391.
Maguire AM, Russell S, Wellman JA, Chung DC, Yu ZF, Tillman A, et al. Efficacy, Safety, and Durability of Voretigene Neparvovec-rzyl in RPE65 Mutation-Associated Inherited Retinal Dystrophy: Results of Phase 1 and 3 Trials. Ophthalmology. 2019;126(9):1273-1285. doi:10.1016/j.ophtha.2019.06.017. PMID:31443789.
Fujinami K, Akiyama K, Tsunoda K, et al. Efficacy and Safety of Voretigene Neparvovec in RPE65-Retinopathy: Results of a Phase III Trial in Japan. Ophthalmol Sci. 2025;5:100876. doi:10.1016/j.xops.2025.100876. PMID:40910102; PMCID:PMC12405627.
Gange WS, Sisk RA, Besirli CG, Lee TC, Havunjian M, Schwartz H, et al. Perifoveal Chorioretinal Atrophy after Subretinal Voretigene Neparvovec-rzyl for RPE65-Mediated Leber Congenital Amaurosis. Ophthalmology. Retina. 2022;6(1):58-64. doi:10.1016/j.oret.2021.03.016. PMID:33838313; PMCID:PMC8497635.
Fischer MD, Simonelli F, Sahni J, Holz FG, Maier R, Fasser C, et al. Real-World Safety and Effectiveness of Voretigene Neparvovec: Results up to 2 Years from the Prospective, Registry-Based PERCEIVE Study. Biomolecules. 2024;14(1). doi:10.3390/biom14010122. PMID:38254722; PMCID:PMC10813228.
Copia il testo dell'articolo e incollalo nell'assistente IA che preferisci.
Articolo copiato negli appunti
Apri un assistente IA qui sotto e incolla il testo copiato nella chat.