Bệnh mù bẩm sinh Leber (Leber congenital amaurosis; LCA) là dạng loạn dưỡng võng mạc bẩm sinh nặng nhất, gây suy giảm thị lực nghiêm trọng từ khi sinh ra đến giai đoạn trẻ nhỏ. Đây là nguyên nhân chính gây suy giảm thị lực ở trẻ em và được biết đến như một nguyên nhân phổ biến của mù bẩm sinh. Biểu hiện lâm sàng đa dạng.
Lần đầu tiên được báo cáo vào năm 1869 bởi bác sĩ nhãn khoa người Đức Theodor Karl Gustav von Leber (1840–1917). Lưu ý rằng bệnh thần kinh thị giác di truyền Leber (LHON) do cùng một Leber báo cáo vào năm 1871 là một bệnh ty thể khởi phát ở độ tuổi 20 và hoàn toàn khác với LCA. Năm 1957, sự biến mất của sóng trên ERG được xác nhận là đặc điểm chung trong chẩn đoán LCA, từ đó tên bệnh được thiết lập.
Tỷ lệ hiện mắc ước tính khi sinh là 2–3 trên 100.000 ca sinh (1/30.000 đến 1/81.000)1). Một số báo cáo đề cập đến 1:80.000 đến 1:200.000, cho thấy sự khác biệt2). LCA chiếm khoảng 5% tổng số các loạn dưỡng võng mạc, và khoảng 20% trẻ em khiếm thị trong các trường dành cho người mù mắc LCA1). Hiện nay, khoảng 27 gen liên quan đến LCA đã được xác định2), và gen gây bệnh được tìm thấy ở khoảng 70–80% trường hợp2). Kiểu di truyền chính là lặn nhiễm sắc thể thường, nhưng cũng có báo cáo về di truyền trội và liên kết nhiễm sắc thể X.
QBệnh mù bẩm sinh Leber có thể được phát hiện khi nào?
A
Thông thường, cha mẹ nhận thấy rung giật nhãn cầu hoặc thiếu khả năng cố định thị giác vào khoảng 6 tuần tuổi3). Nếu có phản ứng thị giác kém (không thể cố định hoặc theo dõi), nghi ngờ LCA và chẩn đoán được xác nhận bằng ERG.
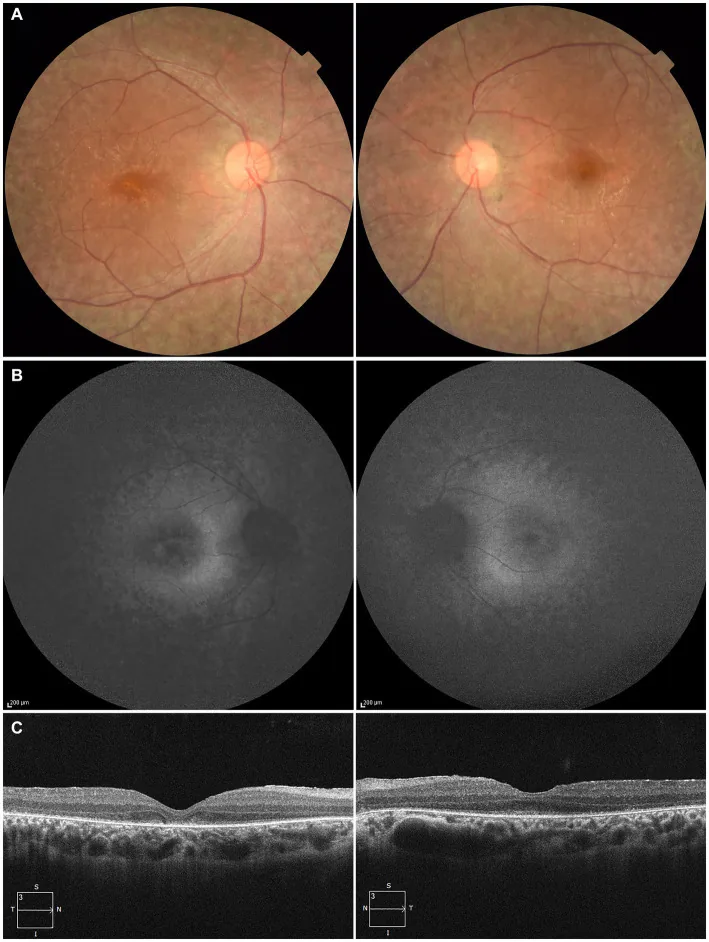
Higa N, et al. A novel RPE65 variant p.(Ala391Asp) in Leber congenital amaurosis: a case report and literature review in Japan. Front Med (Lausanne). 2024. Figure 1. PMCID: PMC11446184. License: CC BY.
Ở bệnh nhân 27 tuổi, (A) ảnh đáy mắt hai mắt cho thấy mất sắc tố biểu mô sắc tố võng mạc và hẹp mạch máu võng mạc, (B) tự huỳnh quang đáy mắt cho thấy tăng huỳnh quang và giảm tín hiệu ở vùng tổn thương, (C) OCT cho thấy mất vùng ellipsoid (EZ) ở mắt phải và còn sót ít ở hố mắt trái. Tương ứng với thoái hóa biểu mô sắc tố võng mạc và hẹp mạch máu võng mạc được đề cập trong phần “2. Triệu chứng chính và dấu hiệu lâm sàng”.
Suy giảm thị lực nghiêm trọng đã có từ khi sinh ra hoặc giai đoạn sớm sau sinh.
Giảm thị lực: Hầu hết bệnh nhân có thị lực dưới 0,1, và khoảng một phần ba không có cảm nhận ánh sáng. Suy giảm thị lực thường ổn định hoặc tiến triển rất chậm.
Sợ ánh sáng: Nhiều trường hợp có biểu hiện quá mẫn với ánh sáng.
Quáng gà: Chức năng thị giác ở nơi tối giảm hơn nữa.
Cha mẹ thường nhận thấy rung giật nhãn cầu hoặc thiếu cố định thị giác vào khoảng 6 tuần tuổi3).
Dấu hiệu lâm sàng (dấu hiệu bác sĩ xác nhận khi khám)
Rung giật nhãn cầu: Xuất hiện khi sinh hoặc ngay sau đó. Có dạng lắc lư hoặc lang thang ở mọi vị trí mắt. Đây là điểm phân biệt quan trọng với loạn dưỡng võng mạc nặng khởi phát sớm (EOSRD), vì EOSRD không kèm rung giật nhãn cầu2).
Bất thường phản ứng đồng tử: Phản xạ ánh sáng chậm hoặc mất. Được gọi là “đồng tử hắc võng mạc (amaurotic pupils)”.
Dấu hiệu mắt-ngón tay (oculo-digital sign): Động tác chọc, ấn hoặc dụi mắt, được cho là cố gắng kích thích cơ học võng mạc để gợi thị giác. Di chứng chính là lõm mắt do teo mỡ hốc mắt.
Tật khúc xạ: Viễn thị nặng (>5 đi-ốp) phổ biến, được cho là do rối loạn quá trình chính thị hóa vì suy giảm thị lực sớm.
Ở giai đoạn trẻ sơ sinh, đáy mắt thường có vẻ bình thường, nhưng sau đó xuất hiện nhiều dấu hiệu đa dạng. Dấu hiệu đáy mắt thay đổi từ đáy mắt bình thường đến đáy mắt giống viêm võng mạc sắc tố điển hình. Trong trường hợp tiến triển, gai thị trở nên nhợt nhạt, mạch máu rất hẹp, màu sắc tổng thể đáy mắt tối, và phản xạ vòng hoàng điểm biến mất.
Đĩa thị nhợt nhạt và hẹp mạch máu võng mạc
Đáy mắt dạng muối tiêu: thoái hóa biểu mô sắc tố võng mạc với các đốm trắng nhỏ và mất sắc tố
Lắng đọng sắc tố hình gai xương và đốm dưới võng mạc (đáy mắt cẩm thạch)
Trên OCT, các lớp ngoài của võng mạc gần như biến mất, với đặc điểm là mỏng đi đến mất vùng ellipsoid (đường nối IS/OS). Kết quả khác nhau tùy theo kiểu gen:
Đột biến CRB1: tăng độ dày võng mạc nghịch thường (phân lớp thô) 1)
LCA được phân loại thành thể đơn thuần chỉ có triệu chứng mắt và thể phức tạp kèm bệnh toàn thân.
Thể đơn giản
Chỉ có triệu chứng mắt: Rối loạn thị giác là chủ yếu, không kèm bất thường toàn thân.
Viễn thị trung bình đến nặng: Thường gặp ở các trường hợp không có bất thường toàn thân.
Thể phức tạp
Bất thường hệ thần kinh trung ương: Thiểu sản thùy nhộng tiểu não, dị dạng thân não.
Chậm phát triển tâm thần: Một số trường hợp kèm khuyết tật trí tuệ.
Bệnh thận: Có thể kèm bệnh thận đa nang.
Khác: Mất thính lực, bất thường xương, bệnh gan, rối loạn chuyển hóa, động kinh.
QMục đích của dấu hiệu dụi mắt (động tác dụi mắt) là gì?
A
Hành vi đặc trưng của bệnh nhân LCA, họ chọc hoặc ấn vào mắt bằng ngón tay để kích thích cơ học võng mạc và cố gắng gợi lại thị giác. Lặp lại lâu dài gây teo mỡ hốc mắt, dẫn đến lõm mắt.
LCA là một nhóm bệnh thoái hóa võng mạc di truyền, hầu hết theo kiểu di truyền lặn nhiễm sắc thể thường2). Hiếm khi, các trường hợp trội nhiễm sắc thể thường xảy ra do đột biến ở CRX, IMPDH1 và OTX22). Cũng có báo cáo về di truyền liên kết X.
Hiện nay, 19 thể LCA1 đến LCA19 đã được báo cáo, cùng với 8 gen liên quan khác2). Các gen gây bệnh tham gia vào nhiều con đường liên quan đến sự phát triển và chức năng võng mạc, như hình thái tế bào cảm quang, lông chuyển dẫn truyền ánh sáng và chu trình thị giác.
Các gen liên quan đến LCA được phân loại thành 5 mạng chức năng chính2):
Chuyển hóa retinoid và chu trình thị giác tế bào que (RPE65, LRAT, RDH12)
Duy trì cân bằng nội môi võng mạc và duy trì tế bào cảm quang (AIPL1, SPATA7, TULP1, USP45, CRB1, LCA5)
Phát triển võng mạc và hình thái học (RD3, CEP290)
Phát hiện kích thích ánh sáng và nhận thức thị giác (GUCY2D, CNGA3)
Lông mao kết nối tế bào cảm quang và duy trì đoạn ngoài (CEP290, RPGRIP1, RPGR)
Các gen đột biến thường gặp nhất trên thế giới và tỷ lệ của chúng như sau:
Gen
Tỷ lệ
Con đường liên quan
CEP290
Khoảng 15%
Chức năng lông mao
GUCY2D
Khoảng 12%
Dẫn truyền tín hiệu ánh sáng (tổng hợp cGMP)
CRB1
Khoảng 10%
Duy trì tính phân cực tế bào
RPE65
Khoảng 8%
Chuyển hóa retinoid
Trong một nhóm thuần tập người Nhật (34 gia đình), phân tích NGS cho thấy tỷ lệ phát hiện khoảng 56%, và các gen đột biến phổ biến nhất được báo cáo là CRB1, NMNAT1 và RPGRIP11).
Việc xác định gen gây bệnh liên quan trực tiếp đến việc đánh giá đủ điều kiện điều trị. Đặc biệt, xác nhận đột biến RPE65 là cần thiết để đánh giá đủ điều kiện cho liệu pháp gen (voretigene neparvovec)2).
AIPL1 hoạt động như một chaperone đặc hiệu cho phosphodiesterase 6 (PDE6), enzyme phân hủy cGMP trong quá trình truyền tín hiệu ánh sáng. Thiếu hụt AIPL1 dẫn đến giảm mạnh lượng protein PDE6, gây rối loạn chuyển hóa cGMP → thoái hóa tế bào cảm thụ ánh sáng → mù lòa sớm2). Đột biến AIPL1 chiếm khoảng 5-10% tổng số ca LCA2).
QXác suất đứa con tiếp theo mắc LCA là bao nhiêu?
A
Trong di truyền lặn trên nhiễm sắc thể thường, nếu cả bố và mẹ đều là người mang gen, xác suất đứa con tiếp theo mắc bệnh là 25%, xác suất là người mang gen là 50%, và xác suất không mắc bệnh và không mang gen là 25%. Nếu gen gây bệnh đã được xác định, có thể cân nhắc chẩn đoán trước sinh hoặc chẩn đoán tiền làm tổ.
Chẩn đoán LCA được thực hiện trên lâm sàng, cần được xác nhận bằng điện võng mạc và xác nhận di truyền phân tử bằng xét nghiệm gen2).
Khi có đáp ứng thị giác bẩm sinh kém rõ rệt (thiếu khả năng cố định và bám theo), nghi ngờ LCA. Ở trẻ sơ sinh bị suy giảm thị lực nặng và viễn thị cao, tìm kiếm LCA bằng xét nghiệm di truyền phân tử là lựa chọn đầu tiên4).
Các xét nghiệm chẩn đoán chính được trình bày dưới đây.
Khác nhau tùy phân nhóm (loại RPE65: mất, loại GUCY2D: bình thường).
Xét nghiệm di truyền (NGS, v.v.)
Cần thiết để chẩn đoán xác định và xác định phân nhóm.
Điện võng mạc: Đáp ứng của tế bào que và tế bào nón đều không có hoặc giảm rõ rệt. ERG bình thường loại trừ chẩn đoán LCA.
OCT: Các lớp ngoài võng mạc và vùng ellipsoid gần như biến mất. Trong đột biến CRB1, có sự gia tăng nghịch lý độ dày võng mạc (phân lớp thô) 1). OCT cầm tay hữu ích để kiểm tra trẻ sơ sinh tỉnh táo hoặc trẻ nhỏ dưới gây mê 4).
FAF: Kết quả khác nhau tùy phân nhóm. Trong đột biến GUCY2D, huỳnh quang tự nhiên bình thường; trong đột biến RPE65, biến mất 2).
Xét nghiệm di truyền: Sử dụng giải trình tự thế hệ mới (NGS), DNA microarray, phân tích liên kết. Tỷ lệ phát hiện tổng thể khoảng 70-80% 2). Từ năm 2023, xét nghiệm bảng gen cho 82 gen gây bệnh IRD (Bảng gen PrismGuide IRD) được bảo hiểm chi trả, áp dụng cho các trường hợp khởi phát sớm nghi ngờ IRD liên quan đến RPE65.
Hầu hết các thể LCA chưa có phương pháp điều trị thực chất. Quản lý hiện tại như sau:
Chỉnh tật khúc xạ: Thực hiện chỉnh khúc xạ phù hợp cho viễn thị nặng, v.v. Có thể có tật khúc xạ lớn, vì vậy hãy kính thuốc và cố gắng tập luyện thị giác.
Tập luyện thị giác: Thực hiện phục hồi chức năng thị giác để tận dụng tối đa chức năng thị giác còn lại.
Tập luyện thay thế thị giác: Do chức năng thị giác bị suy giảm đáng kể, cần cân nhắc hướng dẫn chữ nổi, tập đi với gậy trắng, và sử dụng kính lúp đọc sách.
Tư vấn di truyền: Khuyến nghị cho gia đình và bệnh nhân. Có thể thực hiện xét nghiệm người mang gen, chẩn đoán trước sinh, và chẩn đoán di truyền tiền làm tổ.
Xử trí chứng sợ ánh sáng: Khuyến nghị sử dụng kính chống sáng và giảm tiếp xúc với ánh sáng.
Theo dõi định kỳ: Thực hiện theo dõi nhãn khoa bao gồm điện võng mạc, chuyển đến phòng khám thị lực kém nếu cần.
Năm 2017, FDA Hoa Kỳ đã phê duyệt voretigene neparvovec-rzyl (tên thương mại Luxturna) để điều trị LCA2 liên quan đến đột biến RPE65 hai alen. Đây là sản phẩm liệu pháp gen đầu tiên được FDA phê duyệt trong lĩnh vực nhãn khoa. Năm 2023, thuốc cũng được phê duyệt tại Nhật Bản (tên sản phẩm: Luxturna®注).
Một bản sao bình thường của gen RPE65 được đưa vào biểu mô sắc tố võng mạc thông qua tiêm dưới võng mạc bằng vector virus liên quan đến adeno tái tổ hợp (rAAV2). Quy trình được thực hiện trong phòng phẫu thuật dịch kính.
Thử nghiệm Giai đoạn III 301 (Russell 2017)5):
Đối tượng: 31 bệnh nhân mắc IRD liên quan đến RPE65
Tiêu chí chính: MLMT (kiểm tra khả năng di chuyển đa độ sáng; kiểm tra quan sát hành vi ở các mức độ chiếu sáng khác nhau)
Cũng ghi nhận cải thiện đáng kể ở ngưỡng kích thích toàn trường (FST)
Kết quả dài hạn Giai đoạn I/III (Maguire 2019)6):
Độ nhạy võng mạc, thị lực và lợi ích chức năng đạt đỉnh sau 6-12 tháng điều trị có xu hướng giảm dần sau đó
Sự gia tăng độ nhạy võng mạc có thể cải thiện quáng gà và thị trường
Thử nghiệm Giai đoạn III trong nước (thử nghiệm A11301) (Fujinami 2025)7):
Đối tượng: 4 bệnh nhân Nhật Bản mắc IRD liên quan đến RPE65
Tăng độ nhạy đáng kể ở FST (ngưỡng kích thích toàn trường) (tăng độ nhạy hơn 10 lần được coi là đáng kể)
Mở rộng thị trường được xác nhận tại thời điểm 1 năm sau khi dùng thuốc
Tác dụng phụ chính: rối loạn mắt bao gồm đau mắt (được cho là liên quan đến thủ thuật tiêm)
Phác đồ dùng thuốc:
Mắt thứ hai được tiêm ít nhất 6 ngày sau mắt thứ nhất
Ức chế miễn dịch: bắt đầu steroid 3 ngày trước khi tiêm và tiếp tục trong 14 ngày sau tiêm
Tuy nhiên, đột biến RPE65 chỉ chiếm khoảng 8% tổng số bệnh nhân LCA. Đối với các dạng đột biến khác, hiện chưa có liệu pháp nào được chứng minh hiệu quả.
QLiệu pháp gen có thể dùng cho tất cả bệnh nhân LCA không?
A
Liệu pháp gen hiện được phê duyệt (voretigene neparvovec / Luxturna®) chỉ dành cho LCA2 do đột biến hai alen RPE65. Đột biến RPE65 chỉ chiếm khoảng 8% tổng số LCA, do đó đa số bệnh nhân không đủ điều kiện. Các liệu pháp cho kiểu gen khác vẫn đang trong giai đoạn nghiên cứu.
QLàm thế nào để được điều trị gen tại Nhật Bản?
A
Cần xác nhận đột biến hai alen RPE65 và có đủ tế bào võng mạc sống. Bước đầu tiên là chẩn đoán gen bằng bảng gen (PrismGuide IRD panel). Khuyến nghị chuyển đến cơ sở chuyên khoa có kinh nghiệm trong điều trị bệnh võng mạc di truyền.
Sinh lý bệnh của LCA liên quan đến sự gián đoạn chu trình thị giác (Visual Cycle), khiến mắt không thể truyền thông tin ánh sáng.
Chu trình thị giác là một loạt các phản ứng enzyme giữa biểu mô sắc tố võng mạc (RPE) và võng mạc thần kinh cảm giác, trong đó vitamin A từ chế độ ăn được chuyển hóa để tạo ra 11-cis-retinal, cần thiết để hình thành các sắc tố thị giác. Nếu không có 11-cis-retinal, dòng thác truyền tín hiệu ánh sáng không thể bắt đầu và các tín hiệu thần kinh thị giác không được truyền đến vỏ não thị giác. Đột biến ở bất kỳ gen nào mã hóa protein tham gia vào chuỗi phản ứng này sẽ làm gián đoạn chu trình thị giác và gây ra các triệu chứng LCA.
Về mặt mô bệnh học, có bằng chứng về sự tham gia của võng mạc ngoài và tế bào cảm thụ ánh sáng, cho thấy LCA là một quá trình thoái hóa chứ không phải loạn sản.
GUCY2D (LCA1): Mã hóa guanylate cyclase đặc hiệu võng mạc (GC-E). Xúc tác tổng hợp cGMP, chìa khóa trong dẫn truyền tín hiệu ánh sáng. Hơn 140 đột biến liên quan đến bệnh đã được xác định, 88% gây LCA lặn nhiễm sắc thể thường. Axit amin 838 được biết đến là điểm nóng đột biến 2).
RPE65 (LCA2): Thuộc siêu họ carotenoid cleavage oxygenase, là enzyme hai chức năng xúc tác sự cắt liên kết O-alkyl từ all-trans-retinyl ester và đồng phân hóa phần retinyl (all-trans-retinol → 11-cis-retinol) 2). Cần thiết cho chức năng của cả tế bào que và tế bào nón, các nghiên cứu gần đây gợi ý khả năng tham gia vào quá trình đồng phân hóa lutein thành meso-zeaxanthin 2). Liệu pháp gen duy nhất được phê duyệt.
CRB1 (LCA8): Tương đồng với protein crumbs ở ruồi giấm, được biểu hiện ở đoạn trong của tế bào cảm thụ ánh sáng và tế bào Müller. Quan trọng để duy trì tính phân cực tế bào, nằm trên nhiễm sắc thể 1q31.3 1).
CEP290 (LCA10): Tham gia vào chức năng lông mao của tế bào cảm thụ ánh sáng. Tần số đột biến cao nhất trong số các gen liên quan đến LCA (khoảng 15%).
NMNAT1 (LCA9): Mã hóa enzyme chủ chốt trong sinh tổng hợp NAD (nicotinamide adenine dinucleotide) 2).
LCA5: Mã hóa lebercilin, tham gia vào chức năng lông mao và vận chuyển protein nội lông mao 2).
AIPL1: Hoạt động như một chaperone đặc hiệu cho PDE6 (phosphodiesterase 6). Thiếu hụt AIPL1 dẫn đến mất ổn định PDE6 → rối loạn chuyển hóa cGMP → bất thường kênh → thoái hóa tế bào cảm thụ ánh sáng 2).
7. Nghiên cứu mới nhất và triển vọng tương lai (báo cáo giai đoạn nghiên cứu)
Trong đánh giá theo dõi dài hạn của voretigene neparvovec (NCT00481546, NCT00643747), đã báo cáo đỉnh ban đầu sau 6-12 tháng điều trị, sau đó giảm dần các lợi ích lâm sàng bao gồm độ nhạy võng mạc, thị lực và lợi ích chức năng 6). Nghiên cứu PERCEIVE (nghiên cứu đăng ký tiến cứu) báo cáo dữ liệu an toàn và hiệu quả trong 2 năm trong thực hành lâm sàng thực tế, với viêm màng bồ đào liên quan đến liệu pháp gen (GTAU) được quan sát thấy ở tới 50% trường hợp 9).
Được phát triển bởi Editas Medicine. Vector AAV5 mang Cas9 từ S. aureus và hai RNA dẫn đường, nhắm vào đột biến intron sâu (c.2991+1655A>G) nằm trong intron 26 của CEP290 3). Thử nghiệm first-in-human đã xác nhận tính an toàn và cho thấy khả năng dung nạp tốt ngay cả ở liều tương đối cao 3).
Phục hồi chức năng thị giác bằng Quang di truyền học (Optogenetics)
Là phương pháp tiếp cận không phụ thuộc gen, kỹ thuật biểu hiện channelrhodopsin đáp ứng ánh sáng trên các tế bào thần kinh võng mạc bên trong còn lại đang được nghiên cứu 3). Có khả năng áp dụng cho tất cả các loại LCA (bất kể kiểu gen), và các thử nghiệm lâm sàng ban đầu đang được tiến hành.
Liệu pháp gen cho các đột biến GUCY2D và AIPL1 đang được tiến hành trên mô hình động vật và đã cho thấy kết quả đầy hứa hẹn trong việc cứu sống các tế bào cảm thụ hình que và hình nón.
Diễn tiến của LCA được phân loại thành ba dạng: ổn định (khoảng 75%), xấu đi dần dần (khoảng 15%) và cải thiện (khoảng 10%). Đột biến AIPL1 liên quan đến xấu đi dần dần, trong khi đột biến RPGRIP1 liên quan đến diễn tiến ổn định. Trong tương lai, việc ngăn chặn tiến triển hoặc điều trị bằng thị giác nhân tạo, liệu pháp gen và y học tái tạo đang được kỳ vọng.
Duan W, Zhou T, Jiang H, Zhang M, Hu M, Zhang L. A novel nonsense variant (c.1499C>G) in CRB1 caused Leber congenital amaurosis-8 in a Chinese family and a literature review. BMC Med Genomics. 2022;15(1):197. doi:10.1186/s12920-022-01356-z.
Mordà D, Alibrandi S, Scimone C, Rinaldi C, Scalinci SZ, Abate G, et al. Decoding pediatric inherited retinal dystrophies: Bridging genetic complexity and clinical heterogeneity. Progress in retinal and eye research. 2025;109:101405. doi:10.1016/j.preteyeres.2025.101405. PMID:40998214.
Botto C, Rucli M, Tekinsoy MD, Pulman J, Sahel JA, Dalkara D. Early and late stage gene therapy interventions for inherited retinal degenerations. Progress in retinal and eye research. 2022;86:100975. doi:10.1016/j.preteyeres.2021.100975. PMID:34058340.
Gurnani B, et al. Clinical approach to pediatric nystagmus: a comprehensive diagnostic algorithm. Clin Ophthalmol. 2025;19:1617-1636.
Russell S, Bennett J, Wellman JA, Chung DC, Yu ZF, Tillman A, et al. Efficacy and safety of voretigene neparvovec (AAV2-hRPE65v2) in patients with RPE65-mediated inherited retinal dystrophy: a randomised, controlled, open-label, phase 3 trial. Lancet (London, England). 2017;390(10097):849-860. doi:10.1016/S0140-6736(17)31868-8. PMID:28712537; PMCID:PMC5726391.
Maguire AM, Russell S, Wellman JA, Chung DC, Yu ZF, Tillman A, et al. Efficacy, Safety, and Durability of Voretigene Neparvovec-rzyl in RPE65 Mutation-Associated Inherited Retinal Dystrophy: Results of Phase 1 and 3 Trials. Ophthalmology. 2019;126(9):1273-1285. doi:10.1016/j.ophtha.2019.06.017. PMID:31443789.
Fujinami K, Akiyama K, Tsunoda K, et al. Efficacy and Safety of Voretigene Neparvovec in RPE65-Retinopathy: Results of a Phase III Trial in Japan. Ophthalmol Sci. 2025;5:100876. doi:10.1016/j.xops.2025.100876. PMID:40910102; PMCID:PMC12405627.
Gange WS, Sisk RA, Besirli CG, Lee TC, Havunjian M, Schwartz H, et al. Perifoveal Chorioretinal Atrophy after Subretinal Voretigene Neparvovec-rzyl for RPE65-Mediated Leber Congenital Amaurosis. Ophthalmology. Retina. 2022;6(1):58-64. doi:10.1016/j.oret.2021.03.016. PMID:33838313; PMCID:PMC8497635.
Fischer MD, Simonelli F, Sahni J, Holz FG, Maier R, Fasser C, et al. Real-World Safety and Effectiveness of Voretigene Neparvovec: Results up to 2 Years from the Prospective, Registry-Based PERCEIVE Study. Biomolecules. 2024;14(1). doi:10.3390/biom14010122. PMID:38254722; PMCID:PMC10813228.
Sao chép toàn bộ bài viết và dán vào trợ lý AI bạn muốn dùng.
Đã sao chép bài viết vào clipboard
Mở một trợ lý AI bên dưới và dán nội dung đã sao chép vào ô chat.