Leber congenital amaurosis (LCA) adalah jenis distrofi retina kongenital yang paling berat, menyebabkan gangguan penglihatan berat sejak lahir hingga masa bayi. Penyakit ini merupakan penyebab utama gangguan penglihatan pada anak dan dikenal sebagai penyebab umum kebutaan kongenital. Gambaran klinisnya bervariasi.
Pertama kali dilaporkan pada tahun 1869 oleh dokter mata Jerman Theodor Karl Gustav von Leber (1840–1917). Perlu dicatat bahwa Leber hereditary optic neuropathy (LHON) yang dilaporkan oleh Leber yang sama pada tahun 1871 adalah penyakit mitokondria yang timbul sekitar usia 20 tahun dan sangat berbeda dengan LCA. Pada tahun 1957, hilangnya gelombang pada ERG dikonfirmasi sebagai ciri umum diagnosis LCA, sehingga nama penyakit ini ditetapkan.
Perkiraan prevalensi lahir adalah 2–3 per 100.000 kelahiran (1/30.000 hingga 1/81.000)1). Beberapa laporan menyebutkan 1:80.000 hingga 1:200.000, menunjukkan variasi2). LCA mencakup sekitar 5% dari seluruh distrofi retina, dan sekitar 20% anak tunanetra di sekolah luar biasa untuk tunanetra adalah penderita LCA1). Saat ini sekitar 27 gen terkait LCA telah diidentifikasi2), dan gen penyebab ditemukan pada sekitar 70–80% kasus2). Pola pewarisan utama adalah resesif autosomal, namun ada laporan pewarisan dominan dan terkait kromosom X.
QKapan Leber congenital amaurosis dapat diketahui?
A
Biasanya orang tua menyadari adanya nistagmus atau kurangnya fiksasi pada sekitar usia 6 minggu3). Jika terdapat respons visual yang buruk (tidak mampu fiksasi atau mengikuti objek), LCA dicurigai dan diagnosis ditegakkan dengan ERG.
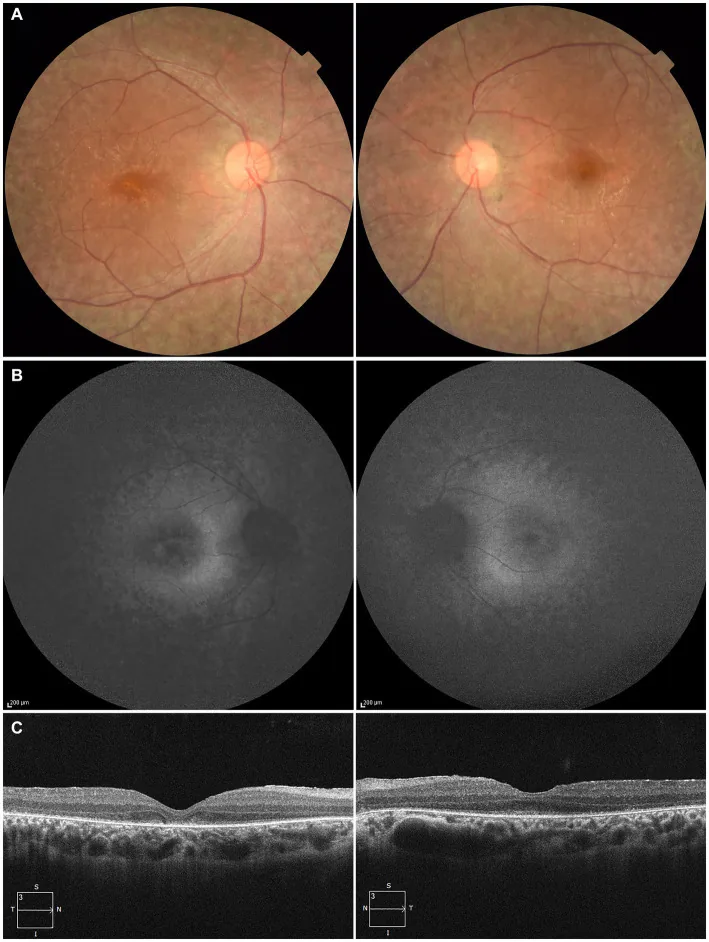
Higa N, et al. A novel RPE65 variant p.(Ala391Asp) in Leber congenital amaurosis: a case report and literature review in Japan. Front Med (Lausanne). 2024. Figure 1. PMCID: PMC11446184. License: CC BY.
Pada pasien berusia 27 tahun, (A) foto fundus kedua mata menunjukkan depigmentasi epitel pigmen retina dan penyempitan pembuluh darah retina, (B) autofluoresensi fundus menunjukkan hiperfluoresensi dan penurunan sinyal pada lesi, (C) OCT menunjukkan hilangnya zona ellipsoid (EZ) pada mata kanan dan sisa sedikit di fovea mata kiri. Sesuai dengan degenerasi epitel pigmen retina dan penyempitan pembuluh darah retina yang dibahas di bagian “2. Gejala utama dan temuan klinis”.
Gangguan penglihatan berat sudah ada sejak lahir atau awal masa postnatal.
Penurunan ketajaman penglihatan: Sebagian besar pasien memiliki ketajaman penglihatan di bawah 0,1, dan sekitar sepertiga tidak memiliki persepsi cahaya. Gangguan penglihatan umumnya stabil atau berkembang sangat lambat.
Fotofobia: Banyak kasus menunjukkan sensitivitas berlebihan terhadap cahaya.
Rabun senja: Fungsi penglihatan di tempat gelap semakin memburuk.
Sering kali orang tua menyadari adanya nistagmus atau kurangnya fiksasi pada sekitar usia 6 minggu3).
Temuan klinis (temuan yang dikonfirmasi dokter saat pemeriksaan)
Nistagmus: Muncul saat lahir atau segera setelahnya. Bersifat pendular atau mengembara di semua posisi mata. Ini merupakan poin diferensial penting dengan distrofi retina berat onset dini (EOSRD), di mana EOSRD tidak disertai nistagmus2).
Kelainan reaksi pupil: Refleks cahaya lambat hingga hilang. Disebut “pupil amaurotik (amaurotic pupils)”.
Tanda okulo-digital (oculo-digital sign): Gerakan menusuk, menekan, atau menggosok mata, dianggap sebagai upaya merangsang retina secara mekanis untuk membangkitkan penglihatan. Sekuela utama adalah enoftalmus akibat atrofi lemak orbita.
Kelainan refraksi: Hipermetropia tinggi (>5 dioptri) umum terjadi, diduga akibat gangguan emetropisasi karena gangguan penglihatan dini.
Pada masa bayi, fundus sering tampak normal, tetapi kemudian muncul berbagai temuan. Temuan fundus bervariasi dari fundus normal hingga fundus seperti retinitis pigmentosa tipikal. Pada kasus lanjut, diskus optikus pucat, pembuluh darah sangat menyempit, warna fundus keseluruhan gelap, dan refleks annular makula menghilang.
Pucatnya diskus optikus dan penyempitan pembuluh darah retina
Fundus garam dan merica: degenerasi epitel pigmen retina dengan bintik putih kecil dan depigmentasi
Pigmentasi seperti duri tulang dan fleck subretina (fundus marmer)
Pada OCT, lapisan luar retina hampir hilang, dengan penipisan hingga hilangnya zona ellipsoid (IS/OS junction). Temuan bervariasi berdasarkan genotipe:
Mutasi CRB1: peningkatan ketebalan retina yang paradoks (coarse lamination) 1)
QApa tujuan dari tanda okular (gerakan menggosok mata)?
A
Perilaku khas pasien LCA, yaitu menusuk atau menekan mata dengan jari untuk merangsang retina secara mekanis dan mencoba membangkitkan penglihatan. Pengulangan jangka panjang menyebabkan atrofi lemak orbita, yang menyebabkan enoftalmus.
LCA adalah kelompok penyakit degenerasi retina herediter, sebagian besar mengikuti pola pewarisan autosomal resesif2). Jarang, kasus autosomal dominan terjadi karena mutasi pada CRX, IMPDH1, dan OTX22). Ada juga laporan pewarisan terkait-X.
Saat ini, 19 tipe LCA1 hingga LCA19 telah dilaporkan, ditambah 8 gen terkait lainnya2). Gen penyebab terlibat dalam beberapa jalur yang berkaitan dengan perkembangan dan fungsi retina, seperti morfogenesis fotoreseptor, silia transduksi cahaya, dan siklus visual.
Gen terkait LCA diklasifikasikan ke dalam 5 jaringan fungsional utama2):
Metabolisme retinoid dan siklus visual batang (RPE65, LRAT, RDH12)
Perkembangan retina dan morfogenesis (RD3, CEP290)
Deteksi rangsangan cahaya dan persepsi visual (GUCY2D, CNGA3)
Silia penghubung fotoreseptor dan pemeliharaan segmen luar (CEP290, RPGRIP1, RPGR)
Gen yang paling sering bermutasi di dunia dan persentasenya adalah sebagai berikut:
Gen
Persentase
Jalur yang terlibat
CEP290
Sekitar 15%
Fungsi silia
GUCY2D
Sekitar 12%
Transduksi sinyal cahaya (sintesis cGMP)
CRB1
Sekitar 10%
Pemeliharaan polaritas sel
RPE65
Sekitar 8%
Metabolisme retinoid
Pada kohort Jepang (34 keluarga), analisis NGS menunjukkan tingkat deteksi sekitar 56%, dan gen yang paling sering bermutasi dilaporkan adalah CRB1, NMNAT1, dan RPGRIP11).
Identifikasi gen penyebab langsung terkait dengan penentuan kelayakan terapi. Khususnya, konfirmasi mutasi RPE65 sangat penting untuk penilaian kelayakan terapi gen (voretigene neparvovec)2).
AIPL1 berfungsi sebagai chaperon khusus untuk fosfodiesterase 6 (PDE6), yang mendegradasi cGMP dalam transduksi sinyal cahaya. Defisiensi AIPL1 menyebabkan penurunan drastis jumlah protein PDE6, yang mengakibatkan gangguan metabolisme cGMP → degenerasi fotoreseptor → kebutaan dini2). Mutasi AIPL1 mencakup sekitar 5-10% dari seluruh kasus LCA2).
QBerapa probabilitas LCA diwariskan kepada anak berikutnya?
A
Pada pewarisan autosomal resesif, jika kedua orang tua adalah karier, probabilitas anak berikutnya terkena adalah 25%, probabilitas menjadi karier adalah 50%, dan probabilitas tidak terkena dan bukan karier adalah 25%. Jika gen penyebab telah diketahui, diagnosis prenatal atau diagnosis preimplantasi dapat menjadi pilihan.
Diagnosis LCA dilakukan secara klinis, dan diperlukan konfirmasi dengan elektroretinografi serta konfirmasi genetik molekuler melalui tes genetik2).
Jika ditemukan respons visual bawaan yang sangat buruk (kurangnya fiksasi dan追随), dicurigai LCA. Pada bayi dengan gangguan penglihatan berat dan hiperopia tinggi, pencarian LCA melalui tes genetik molekuler menjadi pilihan pertama4).
Pemeriksaan diagnostik utama ditunjukkan di bawah ini.
Berbeda menurut subtipe (tipe RPE65: hilang, tipe GUCY2D: normal).
Tes genetik (NGS dll.)
Diperlukan untuk diagnosis pasti dan identifikasi subtipe.
Elektroretinografi: Respons batang dan kerucut keduanya tidak ada atau sangat menurun. ERG normal menyingkirkan diagnosis LCA.
OCT: Lapisan luar retina dan zona ellipsoid hampir hilang. Pada mutasi CRB1, ditemukan peningkatan ketebalan retina yang paradoksal (pelapisan kasar) 1). OCT genggam berguna untuk pemeriksaan bayi yang sadar atau anak kecil yang dibius 4).
FAF: Temuan bervariasi menurut subtipe. Pada mutasi GUCY2D, autofluoresensi normal; pada mutasi RPE65, menghilang 2).
Tes genetik: Digunakan sekuensing generasi berikutnya (NGS), microarray DNA, analisis keterkaitan. Tingkat deteksi keseluruhan sekitar 70-80% 2). Sejak 2023, tes panel untuk 82 gen penyebab IRD (Panel PrismGuide IRD) telah dicakup asuransi, dan diterapkan pada kasus onset dini yang diduga IRD terkait RPE65.
Belum ada terapi substansial untuk sebagian besar jenis LCA. Penanganan saat ini adalah sebagai berikut:
Koreksi kelainan refraksi: Lakukan koreksi refraksi yang tepat untuk hipermetropia berat, dll. Mungkin terdapat kelainan refraksi yang kuat, sehingga berikan resep kacamata dan upayakan pelatihan penglihatan.
Pelatihan penglihatan: Lakukan rehabilitasi visual untuk memaksimalkan fungsi penglihatan yang tersisa.
Pelatihan substitusi penglihatan: Karena gangguan fungsi penglihatan yang signifikan, pertimbangkan instruksi Braille, pelatihan berjalan dengan tongkat putih, dan penggunaan alat pembesar bacaan.
Perawatan low vision: Gunakan alat bantu low vision, dukung akses optimal ke kesempatan pendidikan dan pekerjaan.
Konseling genetik: Direkomendasikan untuk keluarga dan pasien. Tes karier, diagnosis prenatal, dan diagnosis genetik preimplantasi mungkin dilakukan.
Penanganan fotofobia: Disarankan penggunaan kacamata pelindung cahaya dan pengurangan paparan cahaya.
Follow-up rutin: Lakukan follow-up oftalmologi termasuk elektroretinogram, rujuk ke klinik low vision jika diperlukan.
Pada tahun 2017, FDA AS menyetujui voretigene neparvovec-rzyl (nama dagang Luxturna) sebagai pengobatan untuk LCA2 yang terkait dengan mutasi RPE65 bialelik. Ini adalah produk terapi gen pertama yang disetujui FDA di bidang oftalmologi. Pada tahun 2023, obat ini juga disetujui di Jepang (nama produk: Luxturna®注).
Salinan normal gen RPE65 dimasukkan ke dalam epitel pigmen retina melalui injeksi subretina menggunakan vektor virus adeno-associated rekombinan (rAAV2). Prosedur dilakukan di ruang operasi vitrektomi.
Uji Fase III 301 (Russell 2017)5):
Subjek: 31 pasien dengan IRD terkait RPE65
Hasil utama: MLMT (uji mobilitas multi-luminansi; tes observasi perilaku pada berbagai tingkat pencahayaan)
Perbaikan signifikan juga diamati pada ambang stimulus medan penuh (FST)
Hasil jangka panjang Fase I/III (Maguire 2019)6):
Sensitivitas retina, ketajaman penglihatan, dan keuntungan fungsional yang mencapai puncak pada 6-12 bulan setelah pengobatan menunjukkan kecenderungan menurun secara progresif setelahnya
Peningkatan sensitivitas retina diharapkan dapat memperbaiki rabun senja dan lapang pandang
Uji Fase III domestik (uji A11301) (Fujinami 2025)7):
Subjek: 4 pasien Jepang dengan IRD terkait RPE65
Peningkatan sensitivitas signifikan pada FST (ambang stimulus medan penuh) (peningkatan sensitivitas lebih dari 10 kali lipat didefinisikan sebagai signifikan)
Perluasan lapang pandang dikonfirmasi pada 1 tahun setelah pemberian
Efek samping utama: gangguan mata termasuk nyeri mata (diduga terkait dengan prosedur pemberian)
Protokol pemberian:
Mata kedua diberikan setidaknya 6 hari setelah mata pertama
Imunosupresi: steroid dimulai 3 hari sebelum pemberian dan dilanjutkan selama 14 hari setelah pemberian
Namun, mutasi RPE65 hanya mencakup sekitar 8% dari seluruh pasien LCA. Untuk tipe mutasi lainnya, saat ini belum ada terapi yang terbukti efektif.
QApakah terapi gen dapat digunakan untuk semua pasien LCA?
A
Terapi gen yang saat ini disetujui (voretigene neparvovec / Luxturna®) hanya diindikasikan untuk LCA2 akibat mutasi bialelik RPE65. Mutasi RPE65 hanya sekitar 8% dari seluruh LCA, sehingga sebagian besar pasien tidak memenuhi syarat. Terapi untuk genotipe lain masih dalam tahap penelitian.
QBagaimana cara mendapatkan terapi gen di Jepang?
A
Syaratnya adalah konfirmasi mutasi bialelik RPE65 dan adanya sel retina yang masih hidup dalam jumlah cukup. Langkah pertama adalah diagnosis genetik menggunakan panel gen (PrismGuide IRD panel). Disarankan untuk dirujuk ke fasilitas spesialis yang berpengalaman dalam menangani penyakit retina herediter.
6. Patofisiologi dan mekanisme terjadinya secara rinci
Patofisiologi LCA terkait dengan gangguan siklus visual (Visual Cycle), yang menyebabkan mata tidak dapat mentransmisikan informasi cahaya.
Siklus visual adalah serangkaian reaksi enzimatik antara epitel pigmen retina (RPE) dan retina neurosensori, di mana vitamin A dari makanan dimetabolisme untuk menghasilkan 11-cis retinal, yang diperlukan untuk membentuk pigmen visual. Tanpa 11-cis retinal, kaskade transduksi sinyal cahaya tidak dapat dimulai, dan sinyal saraf visual tidak dapat ditransmisikan ke korteks visual. Mutasi pada salah satu gen yang mengkode protein yang terlibat dalam rangkaian reaksi ini akan menghambat siklus visual dan menyebabkan gejala LCA.
Secara histopatologis, terdapat bukti keterlibatan retina luar dan fotoreseptor, menunjukkan bahwa LCA adalah proses degeneratif, bukan displastik.
GUCY2D (LCA1): Mengkode guanilat siklase spesifik retina (GC-E). Mengkatalisis sintesis cGMP, kunci transduksi sinyal cahaya. Lebih dari 140 mutasi terkait penyakit telah diidentifikasi, 88% menyebabkan LCA resesif autosomal. Asam amino 838 dikenal sebagai titik panas mutasi 2).
RPE65 (LCA2): Termasuk dalam superfamili karotenoid cleavage oksigenase, enzim bifungsional yang mengkatalisis pemutusan ikatan O-alkil dari all-trans-retinyl ester dan isomerisasi gugus retinil (all-trans-retinol → 11-cis-retinol) 2). Esensial untuk fungsi batang dan kerucut, penelitian terbaru menunjukkan kemungkinan keterlibatan dalam isomerisasi lutein menjadi meso-zeaxanthin 2). Satu-satunya terapi gen yang disetujui.
CRB1 (LCA8): Homolog dengan protein crumbs pada lalat buah, diekspresikan di segmen dalam fotoreseptor dan sel Müller. Penting untuk pemeliharaan polaritas sel, terletak di kromosom 1q31.3 1).
CEP290 (LCA10): Terlibat dalam fungsi silia fotoreseptor. Frekuensi mutasi tertinggi di antara gen terkait LCA (sekitar 15%).
NMNAT1 (LCA9): Mengkode enzim kunci dalam biosintesis NAD (nikotinamida adenina dinukleotida) 2).
LCA5: Mengkode lebercilin, terlibat dalam fungsi silia dan transportasi protein intrasiliar 2).
AIPL1: Berfungsi sebagai chaperon khusus untuk PDE6 (fosfodiesterase 6). Defisiensi AIPL1 menyebabkan ketidakstabilan PDE6 → gangguan metabolisme cGMP → kelainan saluran → degenerasi fotoreseptor 2).
7. Penelitian terbaru dan prospek masa depan (laporan tahap penelitian)
Dalam evaluasi jangka panjang voretigene neparvovec (NCT00481546, NCT00643747), dilaporkan adanya puncak awal pada 6-12 bulan setelah pengobatan, diikuti penurunan progresif manfaat klinis termasuk sensitivitas retina, ketajaman visual, dan perolehan fungsional 6). Studi PERCEIVE (studi registrasi prospektif) melaporkan data keamanan dan efektivitas 2 tahun dalam praktik klinis nyata, dengan uveitis terkait terapi gen (GTAU) diamati pada hingga 50% kasus 9).
Dikembangkan oleh Editas Medicine. Vektor AAV5 membawa Cas9 dari S. aureus dan dua RNA pemandu, menargetkan mutasi intron dalam (c.2991+1655A>G) yang terletak di intron 26 CEP290 3). Uji coba first-in-human mengonfirmasi keamanan dan menunjukkan tolerabilitas yang baik bahkan pada dosis yang relatif tinggi 3).
Sebagai pendekatan independen gen, teknik mengekspresikan channelrhodopsin yang responsif cahaya pada neuron retina dalam yang tersisa sedang diteliti 3). Berpotensi dapat diterapkan pada semua jenis LCA (tanpa memandang genotipe), dan uji klinis awal sedang berlangsung.
Terapi gen untuk mutasi GUCY2D dan AIPL1 sedang berlangsung pada model hewan, dan telah menunjukkan hasil yang menjanjikan dalam penyelamatan fotoreseptor batang dan kerucut.
Perjalanan LCA diklasifikasikan menjadi tiga pola: stabil (sekitar 75%), memburuk secara progresif (sekitar 15%), dan membaik (sekitar 10%). Mutasi AIPL1 terkait dengan perburukan progresif, sedangkan mutasi RPGRIP1 terkait dengan perjalanan stabil. Di masa depan, penghentian perkembangan atau pengobatan melalui penglihatan buatan, terapi gen, dan pengobatan regeneratif diharapkan.
Duan W, Zhou T, Jiang H, Zhang M, Hu M, Zhang L. A novel nonsense variant (c.1499C>G) in CRB1 caused Leber congenital amaurosis-8 in a Chinese family and a literature review. BMC Med Genomics. 2022;15(1):197. doi:10.1186/s12920-022-01356-z.
Mordà D, Alibrandi S, Scimone C, Rinaldi C, Scalinci SZ, Abate G, et al. Decoding pediatric inherited retinal dystrophies: Bridging genetic complexity and clinical heterogeneity. Progress in retinal and eye research. 2025;109:101405. doi:10.1016/j.preteyeres.2025.101405. PMID:40998214.
Botto C, Rucli M, Tekinsoy MD, Pulman J, Sahel JA, Dalkara D. Early and late stage gene therapy interventions for inherited retinal degenerations. Progress in retinal and eye research. 2022;86:100975. doi:10.1016/j.preteyeres.2021.100975. PMID:34058340.
Gurnani B, et al. Clinical approach to pediatric nystagmus: a comprehensive diagnostic algorithm. Clin Ophthalmol. 2025;19:1617-1636.
Russell S, Bennett J, Wellman JA, Chung DC, Yu ZF, Tillman A, et al. Efficacy and safety of voretigene neparvovec (AAV2-hRPE65v2) in patients with RPE65-mediated inherited retinal dystrophy: a randomised, controlled, open-label, phase 3 trial. Lancet (London, England). 2017;390(10097):849-860. doi:10.1016/S0140-6736(17)31868-8. PMID:28712537; PMCID:PMC5726391.
Maguire AM, Russell S, Wellman JA, Chung DC, Yu ZF, Tillman A, et al. Efficacy, Safety, and Durability of Voretigene Neparvovec-rzyl in RPE65 Mutation-Associated Inherited Retinal Dystrophy: Results of Phase 1 and 3 Trials. Ophthalmology. 2019;126(9):1273-1285. doi:10.1016/j.ophtha.2019.06.017. PMID:31443789.
Fujinami K, Akiyama K, Tsunoda K, et al. Efficacy and Safety of Voretigene Neparvovec in RPE65-Retinopathy: Results of a Phase III Trial in Japan. Ophthalmol Sci. 2025;5:100876. doi:10.1016/j.xops.2025.100876. PMID:40910102; PMCID:PMC12405627.
Gange WS, Sisk RA, Besirli CG, Lee TC, Havunjian M, Schwartz H, et al. Perifoveal Chorioretinal Atrophy after Subretinal Voretigene Neparvovec-rzyl for RPE65-Mediated Leber Congenital Amaurosis. Ophthalmology. Retina. 2022;6(1):58-64. doi:10.1016/j.oret.2021.03.016. PMID:33838313; PMCID:PMC8497635.
Fischer MD, Simonelli F, Sahni J, Holz FG, Maier R, Fasser C, et al. Real-World Safety and Effectiveness of Voretigene Neparvovec: Results up to 2 Years from the Prospective, Registry-Based PERCEIVE Study. Biomolecules. 2024;14(1). doi:10.3390/biom14010122. PMID:38254722; PMCID:PMC10813228.
Salin teks artikel dan tempelkan ke asisten AI pilihan Anda.
Artikel disalin ke papan klip
Buka asisten AI di bawah, lalu tempelkan teks yang disalin ke kotak chat.