L’amaurose congénitale de Leber (LCA) est la forme la plus sévère des dystrophies rétiniennes congénitales, entraînant un handicap visuel sévère dès la naissance ou la petite enfance. C’est une cause majeure de déficience visuelle chez l’enfant et une cause représentative de cécité congénitale. Le tableau clinique est varié.
Elle a été décrite pour la première fois en 1869 par l’ophtalmologiste allemand Theodor Karl Gustav von Leber (1840–1917). La neuropathie optique héréditaire de Leber (LHON), rapportée par le même Leber en 1871, est une maladie mitochondriale survenant vers l’âge de 20 ans, totalement différente de la LCA. En 1957, l’absence d’ondes à l’électrorétinogramme (ERG) a été confirmée comme caractéristique commune du diagnostic de la LCA, établissant le nom de la maladie.
La prévalence estimée à la naissance est de 2 à 3 pour 100 000 naissances (1/30 000 à 1/81 000)1). Certaines sources rapportent des chiffres de 1:80 000 à 1:200 000, avec une variabilité2). Elle représente environ 5 % de toutes les dystrophies rétiniennes, et environ 20 % des enfants malvoyants fréquentant des écoles pour aveugles sont atteints de LCA1). Environ 27 gènes associés à la LCA ont été identifiés à ce jour2), et le gène responsable est identifié dans environ 70 à 80 % des cas2). Le mode de transmission est principalement autosomique récessif, mais des cas de transmission dominante et liée à l’X ont été rapportés.
QQuand l'amaurose congénitale de Leber est-elle généralement détectée ?
A
Généralement, les parents remarquent un nystagmus ou une absence de fixation vers l’âge de 6 semaines3). En cas de mauvaise réponse visuelle sévère (absence totale de fixation et de poursuite), on suspecte une LCA, confirmée par électrorétinogramme.
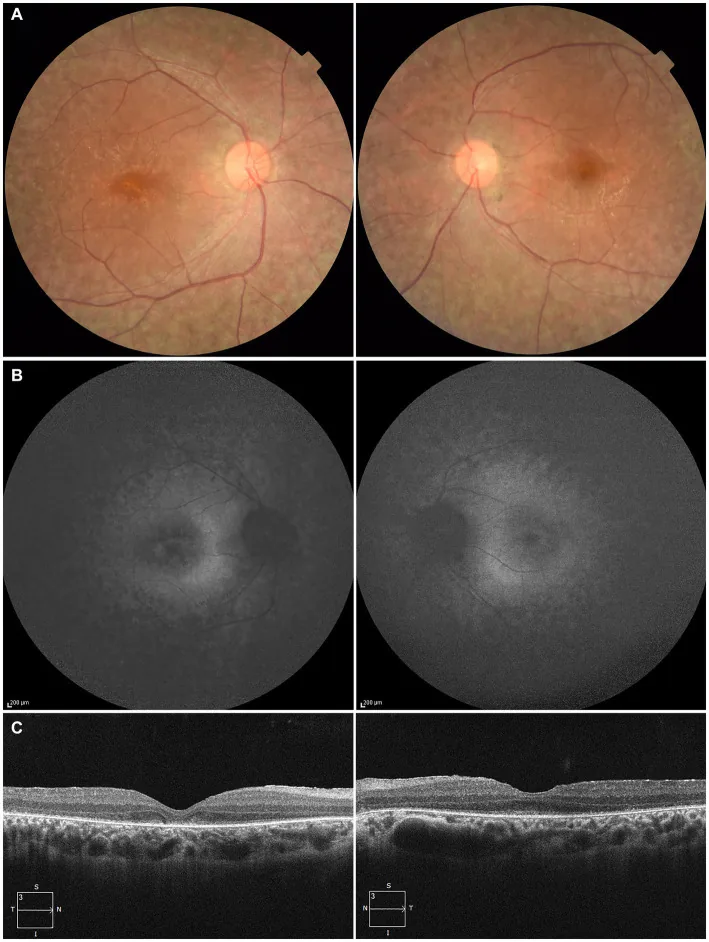
Higa N, et al. A novel RPE65 variant p.(Ala391Asp) in Leber congenital amaurosis: a case report and literature review in Japan. Front Med (Lausanne). 2024. Figure 1. PMCID: PMC11446184. License: CC BY.
Chez un patient de 27 ans, (A) photographies du fond d’œil montrant une dépigmentation de l’épithélium pigmentaire rétinien et un rétrécissement des vaisseaux rétiniens, (B) autofluorescence du fond d’œil montrant une hyperfluorescence et une diminution du signal dans les zones lésées, (C) OCT montrant une disparition de la zone ellipsoïde (EZ) de l’œil droit et une faible persistance au niveau de la fovéa de l’œil gauche. Cela correspond à la dégénérescence de l’épithélium pigmentaire rétinien et au rétrécissement des vaisseaux rétiniens traités dans la section « 2. Principaux symptômes et signes cliniques ».
Une déficience visuelle sévère est présente dès la naissance ou peu après.
Baisse de l’acuité visuelle : La plupart des patients ont une acuité visuelle inférieure à 0,1, et environ un tiers n’ont pas de perception lumineuse. La déficience visuelle est généralement stable ou progresse très lentement.
Photophobie : De nombreux patients présentent une sensibilité à la lumière.
Héméralopie : La fonction visuelle dans l’obscurité est encore plus réduite.
Les parents remarquent souvent un nystagmus ou un défaut de fixation vers l’âge de 6 semaines 3).
Signes cliniques (observés par le médecin lors de l’examen)
Nystagmus : Apparaît à la naissance ou peu après. Il est pendulaire ou erratique et présent dans toutes les positions du regard. C’est un point de différenciation important avec la dystrophie rétinienne sévère à début précoce (EOSRD), qui ne s’accompagne pas de nystagmus2).
Anomalie de la réaction pupillaire : Le réflexe photomoteur est lent ou absent. On parle de « pupilles amaurotiques ».
Signe oculo-digital : Le patient se pique, se pousse ou se frotte les yeux, ce qui est considéré comme une tentative de stimuler mécaniquement la rétine pour évoquer une vision. La principale séquelle est l’enophtalmie due à l’atrophie de la graisse orbitaire.
Erreur de réfraction : Une hypermétropie forte (>5 dioptries) est courante, probablement due à un trouble de l’emmétropisation causé par une déficience visuelle précoce.
Au cours de la petite enfance, le fond d’œil peut souvent sembler normal, mais des signes variés apparaissent ensuite. Les observations du fond d’œil vont d’un fond normal à un aspect typique de rétinite pigmentaire. Dans les cas avancés, la papille optique pâlit, les vaisseaux deviennent très rétrécis, la teinte globale du fond d’œil s’assombrit et le réflexe annulaire maculaire disparaît.
Pâleur de la papille optique et rétrécissement des vaisseaux rétiniens
Fond d’œil poivre et sel : petites taches blanches et dépigmentation de l’épithélium pigmentaire rétinien
Pigmentation en spicules osseux et drusen sous-rétiniens (fond d’œil marbré)
En OCT, les couches externes de la rétine sont presque absentes, avec un amincissement à disparition de la zone ellipsoïde (jonction IS/OS). Les signes varient selon le génotype :
Mutation CRB1 : épaississement paradoxal de la rétine (lamination grossière) 1)
Mutation RPE65 : disparition de l’autofluorescence du fond d’œil (FAF) 2)
La LCA se divise en forme simple (uniquement oculaire) et forme complexe (associée à des maladies systémiques).
Type simple
Symptômes oculaires uniquement : la déficience visuelle est prédominante, sans anomalie systémique.
Hypermétropie modérée à sévère : fréquente dans les cas sans anomalie systémique.
Type complexe
Anomalies du système nerveux central : hypoplasie du vermis cérébelleux, malformations du tronc cérébral.
Retard du développement mental : certains cas présentent une déficience intellectuelle.
Insuffisance rénale : peut être associée à une polykystose rénale.
Autres : surdité, anomalies squelettiques, troubles hépatiques, troubles métaboliques, épilepsie.
QÀ quoi sert le signe oculo-digital (frottement des yeux) ?
A
C’est un comportement caractéristique des patients atteints de LCA : ils se frottent ou se pressent les yeux avec les doigts pour stimuler mécaniquement la rétine et tenter de provoquer une perception visuelle. La répétition à long terme entraîne une atrophie de la graisse orbitaire et peut provoquer une énophtalmie.
La LCA est un groupe de maladies héréditaires de dégénérescence rétinienne, dont la majorité suit un mode de transmission autosomique récessif2). Rarement, des mutations de CRX, IMPDH1 ou OTX2 peuvent entraîner une transmission autosomique dominante2). Des cas de transmission liée à l’X ont également été rapportés.
Actuellement, 19 types de LCA (LCA1 à LCA19) ont été identifiés, ainsi que 8 gènes associés supplémentaires2). Les gènes responsables sont impliqués dans plusieurs voies du développement et du fonctionnement de la rétine, notamment la morphogenèse des photorécepteurs, le cil de transmission de l’information lumineuse et le cycle visuel.
Les gènes associés à la LCA sont classés en cinq principaux réseaux fonctionnels2) :
Métabolisme des rétinoïdes et cycle visuel des bâtonnets (RPE65, LRAT, RDH12)
Maintien de l’homéostasie rétinienne et des photorécepteurs (AIPL1, SPATA7, TULP1, USP45, CRB1, LCA5)
Développement et morphogenèse rétinienne (RD3, CEP290)
Détection de la lumière et perception visuelle (GUCY2D, CNGA3)
Cils connecteurs des photorécepteurs et maintien du segment externe (CEP290, RPGRIP1, RPGR)
Les gènes mutés les plus fréquents dans le monde et leurs proportions sont les suivants :
Gène
Proportion
Voie impliquée
CEP290
Environ 15 %
Fonction ciliaire
GUCY2D
Environ 12 %
Transmission du signal lumineux (synthèse du cGMP)
CRB1
Environ 10 %
Maintien de la polarité cellulaire
RPE65
environ 8 %
Métabolisme des rétinoïdes
Dans une cohorte japonaise (34 familles), l’analyse NGS a montré un taux de détection d’environ 56 %, les gènes mutés les plus fréquents étant CRB1, NMNAT1 et RPGRIP11).
L’identification du gène causal est directement liée à la détermination de l’éligibilité au traitement. En particulier, la confirmation d’une mutation RPE65 est essentielle pour évaluer l’éligibilité à la thérapie génique (vorétigène néparvovec)2).
AIPL1 fonctionne comme un chaperon spécifique de la phosphodiestérase 6 (PDE6), qui dégrade le GMPc dans la transduction du signal lumineux. La déficience en AIPL1 entraîne une diminution drastique de la protéine PDE6, conduisant à une perturbation du métabolisme du GMPc, une dégénérescence des photorécepteurs et une cécité précoce2). Les mutations d’AIPL1 représentent environ 5 à 10 % de toutes les LCA2).
QQuelle est la probabilité que le prochain enfant hérite de la LCA ?
A
Dans le cas d’une transmission autosomique récessive, si les deux parents sont porteurs, le risque pour le prochain enfant d’être atteint est de 25 %, d’être porteur de 50 %, et d’être non atteint et non porteur de 25 %. Si le gène causal est identifié, un diagnostic prénatal ou un diagnostic préimplantatoire est possible.
Le diagnostic de la LCA est clinique, et nécessite une confirmation par électrorétinographie et une confirmation moléculaire par test génétique2).
La LCA est suspectée en cas de réponse visuelle sévèrement réduite dès la naissance (absence de fixation et de poursuite). Chez les nourrissons présentant une déficience visuelle sévère et une hypermétropie élevée, la recherche de LCA par test génétique moléculaire est de première intention4).
Les principaux examens diagnostiques sont présentés ci-dessous.
Variable selon le sous-type (disparition dans le type RPE65, normale dans le type GUCY2D).
Test génétique (NGS, etc.)
Nécessaire pour le diagnostic définitif et l’identification du sous-type.
Électrorétinographie : Les réponses des bâtonnets et des cônes sont absentes à fortement diminuées. Un ERG normal exclut le diagnostic de LCA.
OCT : Les couches externes de la rétine et la zone ellipsoïde sont presque absentes. Les mutations CRB1 montrent un épaississement rétinien paradoxal (lamination grossière)1). L’OCT portable est utile pour examiner les nourrissons éveillés ou les jeunes enfants sous anesthésie4).
FAF : Les résultats varient selon le sous-type. Les mutations GUCY2D conservent une autofluorescence normale, tandis que les mutations RPE65 la font disparaître2).
Test génétique : Le séquençage de nouvelle génération (NGS), les puces à ADN, l’analyse de liaison, etc. sont utilisés. Le taux de détection global est d’environ 70 à 80 %2). Depuis 2023, un test panel pour 82 gènes pathogènes de la RD (panel PrismGuide IRD) est pris en charge par l’assurance maladie et s’applique aux jeunes patients suspectés de RD liée à RPE65.
Pour la plupart des formes d’ALC, aucun traitement curatif n’est établi. La prise en charge actuelle est la suivante :
Correction des erreurs de réfraction : Correction appropriée de l’hypermétropie forte, etc. Une forte erreur de réfraction peut être présente, donc prescrire des lunettes et encourager l’orthoptie.
Orthoptie : Réadaptation visuelle pour utiliser au maximum la fonction visuelle résiduelle.
Entraînement aux aides visuelles : En raison d’une altération significative de la fonction visuelle, envisager l’apprentissage du braille, la formation à la canne blanche, l’utilisation de loupes électroniques, etc.
Soins basse vision : Utilisation d’aides basse vision, soutien pour un accès optimal à l’éducation et à l’emploi.
Conseil génétique : Recommandé pour les familles et les patients. Tests de porteur, diagnostic prénatal, diagnostic préimplantatoire peuvent être possibles.
Gestion de la photophobie : Utilisation de lunettes filtrantes et réduction de l’exposition à la lumière recommandées.
Suivi régulier : Suivi ophtalmologique incluant électrorétinogramme, orientation vers une consultation basse vision si nécessaire.
En 2017, la FDA américaine a approuvé le vorétigène néparvovec-rzyl (nom commercial Luxturna) comme traitement de la LCA2 associée à des mutations bialléliques de RPE65. Il s’agit du premier produit de thérapie génique approuvé par la FDA dans le domaine ophtalmique. En 2023, il a également été approuvé au Japon (nom commercial : Luxturna®).
Un vecteur adéno-associé recombinant (rAAV2) est utilisé pour introduire une copie normale du gène RPE65 dans l’épithélium pigmentaire rétinien par injection sous-rétinienne. L’intervention est réalisée en salle de vitrectomie.
Essai de phase III 301 (Russell 2017)5) :
Sujets : 31 patients atteints de IRD liée à RPE65
Critère principal : MLMT (test de mobilité multi-luminance)
Amélioration significative également observée au test de seuil de stimulation du champ visuel total (FST)
Résultats à long terme de phase I/III (Maguire 2019)6) :
La sensibilité rétinienne, l’acuité visuelle et les gains fonctionnels, qui atteignent un pic 6 à 12 mois après le traitement, ont montré une tendance à diminuer progressivement par la suite.
L’augmentation de la sensibilité rétinienne peut améliorer la cécité nocturne et le champ visuel.
Essai de phase III national (A11301) (Fujinami 2025)7) :
Sujets : 4 patients japonais atteints de IRD liée à RPE65
Augmentation significative de la sensibilité au FST (définie comme une augmentation de la sensibilité de plus de 10 fois)
Élargissement du champ visuel confirmé à 1 an après l’administration.
Principaux événements indésirables : troubles oculaires incluant douleur oculaire (supposés liés à la procédure d’administration)
Protocole d’administration :
Le deuxième œil est traité au moins 6 jours après le premier
Immunosuppression : début des stéroïdes 3 jours avant l’administration, poursuite pendant 14 jours après
Cependant, les mutations RPE65 ne représentent qu’environ 8 % de l’ensemble des patients atteints de LCA. Pour les autres types de mutations, il n’existe actuellement aucun traitement dont l’efficacité a été prouvée.
QLa thérapie génique est-elle utilisable pour tous les patients atteints de LCA ?
A
Actuellement, la thérapie génique approuvée (vorétigène néparvovec / Luxturna®) est indiquée uniquement pour la LCA2 due à des mutations bialléliques de RPE65. Les mutations RPE65 représentent environ 8 % de l’ensemble des LCA, et la majorité des patients n’ont pas cette indication. Les traitements pour d’autres génotypes sont en phase de recherche.
QComment recevoir une thérapie génique au Japon ?
A
Les conditions sont la confirmation d’une mutation biallélique de RPE65 et la présence de cellules rétiniennes viables suffisantes. Le diagnostic génétique par panel de gènes (PrismGuide IRD panel) est la première étape. Une orientation vers un centre spécialisé ayant une expérience dans la prise en charge des maladies rétiniennes héréditaires est recommandée.
La physiopathologie de la LCA est liée à la perturbation du cycle visuel, empêchant l’œil de transmettre les informations lumineuses.
Le cycle visuel est une série de réactions enzymatiques entre l’épithélium pigmentaire rétinien (EPR) et la rétine neurosensorielle, qui métabolise la vitamine A d’origine alimentaire pour produire du 11-cis-rétinal et générer des pigments photorécepteurs. Sans 11-cis-rétinal, la cascade de transduction du signal lumineux ne peut pas démarrer, et les signaux nerveux visuels ne sont pas transmis au cortex visuel. Une mutation dans l’un des gènes codant pour les protéines impliquées dans cette série de réactions peut bloquer le cycle visuel et provoquer les symptômes de la LCA.
Sur le plan histopathologique, l’implication de la rétine externe et des photorécepteurs a été démontrée, suggérant que la LCA est un processus dégénératif plutôt qu’une dysplasie.
GUCY2D (LCA1) : code pour la guanylate cyclase spécifique de la rétine (GC-E). Catalyse la synthèse du cGMP, clé de la phototransduction. Plus de 140 mutations pathogènes identifiées, 88% sont responsables de LCA autosomique récessive. L’acide aminé 838 est un point chaud de mutation connu 2).
RPE65 (LCA2) : appartient à la superfamille des caroténoïdes clivant les oxygénases. C’est une enzyme bifonctionnelle catalysant le clivage de l’ester O-alkylé à partir de l’all-trans-rétinyl ester et l’isomérisation du fragment rétinyl (all-trans-rétinol → 11-cis-rétinol) 2). Essentiel pour la fonction des bâtonnets et des cônes. Des études récentes suggèrent un rôle possible dans l’isomérisation de la lutéine en méso-zéaxanthine 2). Seule indication approuvée pour la thérapie génique.
CRB1 (LCA8) : homologue de la protéine crumbs chez la drosophile, exprimé dans le segment interne des photorécepteurs et les cellules de Müller. Important pour le maintien de la polarité cellulaire, situé sur le chromosome 1q31.3 1).
CEP290 (LCA10) : impliqué dans la fonction ciliaire des photorécepteurs. Le gène le plus fréquemment muté parmi les gènes associés à la LCA (environ 15%).
NMNAT1 (LCA9) : code pour une enzyme clé de la biosynthèse du NAD (nicotinamide adénine dinucléotide) 2).
LCA5 : code pour la leberciline, impliquée dans la fonction ciliaire et le transport intraciliaire des protéines 2).
AIPL1 : fonctionne comme un chaperon spécifique de la PDE6 (phosphodiestérase 6). La déficience en AIPL1 déstabilise la PDE6 → perturbation du métabolisme du cGMP → anomalie des canaux → dégénérescence des photorécepteurs 2).
7. Recherches récentes et perspectives futures (rapports en phase de recherche)
L’évaluation de suivi à long terme du vorétigène néparvovec (NCT00481546, NCT00643747) rapporte un pic initial à 6-12 mois après le traitement, suivi d’une diminution progressive des bénéfices cliniques, y compris la sensibilité rétinienne, l’acuité visuelle et les gains fonctionnels 6). L’étude PERCEIVE (étude prospective de registre) a rapporté des données de sécurité et d’efficacité à 2 ans en pratique clinique réelle, avec une uvéite associée à la thérapie génique (GTAU) observée dans jusqu’à 50% des cas 9).
Développé par Editas Medicine. Le vecteur AAV5 transporte Cas9 de S. aureus et deux ARN guides, ciblant la mutation intronique profonde (c.2991+1655A>G) dans l’intron 26 de CEP2903). L’essai first-in-human a confirmé la sécurité et une bonne tolérance même à des doses relativement élevées3).
Restauration de la fonction visuelle par optogénétique
En tant qu’approche indépendante du gène, une méthode consistant à exprimer la channelrhodopsine photosensible dans les neurones rétiniens internes résiduels est étudiée3). Elle pourrait être applicable à tous les types de LCA (indépendamment du génotype) et des essais cliniques précoces sont en cours.
La thérapie génique pour les mutations GUCY2D et AIPL1 est en cours dans des modèles animaux, montrant des résultats prometteurs pour le sauvetage des photorécepteurs à bâtonnets et à cônes.
L’évolution de la LCA se divise en trois schémas : stable (environ 75 %), aggravation progressive (environ 15 %) et amélioration (environ 10 %). Les mutations AIPL1 sont associées à une aggravation progressive, tandis que les mutations RPGRIP1 sont liées à une évolution stable. À l’avenir, on espère un arrêt de la progression ou un traitement par vision artificielle, thérapie génique ou médecine régénérative.
Duan W, Zhou T, Jiang H, Zhang M, Hu M, Zhang L. A novel nonsense variant (c.1499C>G) in CRB1 caused Leber congenital amaurosis-8 in a Chinese family and a literature review. BMC Med Genomics. 2022;15(1):197. doi:10.1186/s12920-022-01356-z.
Mordà D, Alibrandi S, Scimone C, Rinaldi C, Scalinci SZ, Abate G, et al. Decoding pediatric inherited retinal dystrophies: Bridging genetic complexity and clinical heterogeneity. Progress in retinal and eye research. 2025;109:101405. doi:10.1016/j.preteyeres.2025.101405. PMID:40998214.
Botto C, Rucli M, Tekinsoy MD, Pulman J, Sahel JA, Dalkara D. Early and late stage gene therapy interventions for inherited retinal degenerations. Progress in retinal and eye research. 2022;86:100975. doi:10.1016/j.preteyeres.2021.100975. PMID:34058340.
Gurnani B, et al. Clinical approach to pediatric nystagmus: a comprehensive diagnostic algorithm. Clin Ophthalmol. 2025;19:1617-1636.
Russell S, Bennett J, Wellman JA, Chung DC, Yu ZF, Tillman A, et al. Efficacy and safety of voretigene neparvovec (AAV2-hRPE65v2) in patients with RPE65-mediated inherited retinal dystrophy: a randomised, controlled, open-label, phase 3 trial. Lancet (London, England). 2017;390(10097):849-860. doi:10.1016/S0140-6736(17)31868-8. PMID:28712537; PMCID:PMC5726391.
Maguire AM, Russell S, Wellman JA, Chung DC, Yu ZF, Tillman A, et al. Efficacy, Safety, and Durability of Voretigene Neparvovec-rzyl in RPE65 Mutation-Associated Inherited Retinal Dystrophy: Results of Phase 1 and 3 Trials. Ophthalmology. 2019;126(9):1273-1285. doi:10.1016/j.ophtha.2019.06.017. PMID:31443789.
Fujinami K, Akiyama K, Tsunoda K, et al. Efficacy and Safety of Voretigene Neparvovec in RPE65-Retinopathy: Results of a Phase III Trial in Japan. Ophthalmol Sci. 2025;5:100876. doi:10.1016/j.xops.2025.100876. PMID:40910102; PMCID:PMC12405627.
Gange WS, Sisk RA, Besirli CG, Lee TC, Havunjian M, Schwartz H, et al. Perifoveal Chorioretinal Atrophy after Subretinal Voretigene Neparvovec-rzyl for RPE65-Mediated Leber Congenital Amaurosis. Ophthalmology. Retina. 2022;6(1):58-64. doi:10.1016/j.oret.2021.03.016. PMID:33838313; PMCID:PMC8497635.
Fischer MD, Simonelli F, Sahni J, Holz FG, Maier R, Fasser C, et al. Real-World Safety and Effectiveness of Voretigene Neparvovec: Results up to 2 Years from the Prospective, Registry-Based PERCEIVE Study. Biomolecules. 2024;14(1). doi:10.3390/biom14010122. PMID:38254722; PMCID:PMC10813228.
Copiez le texte de l'article et collez-le dans l'assistant IA de votre choix.
Article copié dans le presse-papiers
Ouvrez un assistant IA ci-dessous et collez le texte copié dans la conversation.