Врожденный амавроз Лебера (LCA) является наиболее тяжелой формой врожденной дистрофии сетчатки, приводящей к тяжелым нарушениям зрения с рождения или в младенчестве. Это основная причина нарушения зрения у детей и известна как типичная причина врожденной слепоты. Клиническая картина разнообразна.
Впервые описан в 1869 году немецким офтальмологом Теодором Карлом Густавом фон Лебером (1840–1917). Наследственная оптическая нейропатия Лебера (LHON), описанная тем же Лебером в 1871 году, является митохондриальным заболеванием, возникающим около 20 лет, и полностью отличается от LCA. В 1957 году отсутствие волн на электроретинограмме (ЭРГ) было подтверждено как общая характеристика диагностики LCA, и название болезни утвердилось.
Предполагаемая распространенность при рождении составляет 2–3 на 100 000 рождений (1/30 000 – 1/81 000)1). В некоторых источниках указываются цифры от 1:80 000 до 1:200 000, что свидетельствует о вариабельности2). На LCA приходится около 5% всех дистрофий сетчатки, и около 20% детей с нарушениями зрения, посещающих школы для слепых, страдают LCA1). В настоящее время идентифицировано около 27 генов, ассоциированных с LCA2), и примерно в 70–80% случаев выявляется причинный ген2). Тип наследования в основном аутосомно-рецессивный, но имеются сообщения о доминантном и X-сцепленном наследовании.
QКогда обычно выявляется врожденный амавроз Лебера?
A
Обычно родители замечают нистагм или отсутствие фиксации в возрасте около 6 недель3). При тяжелом нарушении зрительной реакции (полное отсутствие фиксации и прослеживания) подозревают LCA, диагноз подтверждается электроретинограммой.
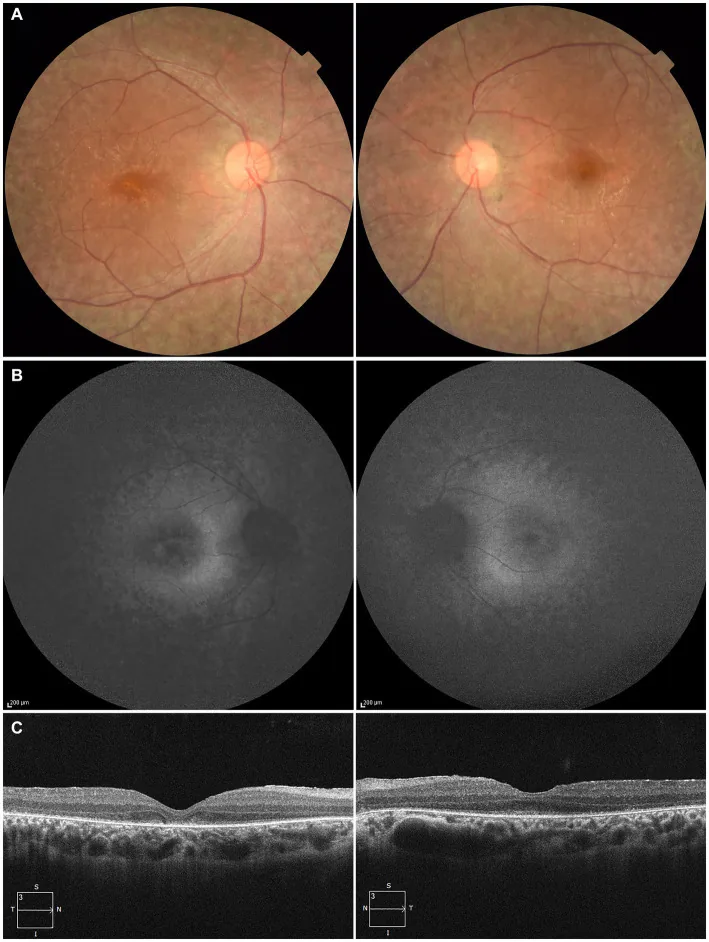
Higa N, et al. A novel RPE65 variant p.(Ala391Asp) in Leber congenital amaurosis: a case report and literature review in Japan. Front Med (Lausanne). 2024. Figure 1. PMCID: PMC11446184. License: CC BY.
У 27-летнего пациента на (A) фотографиях глазного дна обоих глаз видна депигментация пигментного эпителия сетчатки и сужение сосудов сетчатки, (B) аутофлуоресценция глазного дна показывает гиперфлуоресценцию и снижение сигнала в зонах поражения, (C) ОКТ демонстрирует исчезновение эллипсоидной зоны (EZ) на правом глазу и незначительное сохранение в фовеа левого глаза. Это соответствует дегенерации пигментного эпителия сетчатки и сужению сосудов сетчатки, описанным в разделе «2. Основные симптомы и клинические признаки».
Тяжелое нарушение зрения присутствует с рождения или вскоре после рождения.
Снижение остроты зрения: У большинства пациентов острота зрения ниже 0,1, и примерно у трети отсутствует светоощущение. Нарушение зрения обычно стабильно или прогрессирует очень медленно.
Светобоязнь: Многие пациенты проявляют повышенную чувствительность к свету.
Куриная слепота: Зрительная функция в темноте еще больше снижена.
Родители часто замечают нистагм или отсутствие фиксации в возрасте около 6 недель 3).
Клинические признаки (обнаруживаемые врачом при осмотре)
Нистагм: Появляется при рождении или вскоре после него. Он маятникообразный или блуждающий, присутствует во всех положениях глаз. Это важный дифференциальный признак с тяжелой дистрофией сетчатки с ранним началом (EOSRD), которая не сопровождается нистагмом 2).
Нарушение зрачковой реакции: Зрачковый рефлекс на свет вялый или отсутствует. Это называется «амавротические зрачки».
Окуло-дигитальный симптом: Пациент тычет, давит или трет глаза, что рассматривается как попытка механической стимуляции сетчатки для вызывания зрительных ощущений. Основным последствием является энофтальм из-за атрофии орбитального жира.
Аномалия рефракции: Часто встречается высокая гиперметропия (>5 диоптрий), предположительно из-за нарушения эмметропизации вследствие раннего нарушения зрения.
В младенчестве глазное дно часто может выглядеть нормальным, но впоследствии появляются разнообразные изменения. Находки глазного дна варьируют от нормального дна до типичного пигментного ретинита. В запущенных случаях диск зрительного нерва бледнеет, сосуды сильно сужаются, общий тон глазного дна темнеет, и исчезает макулярный кольцевой рефлекс.
Бледность диска зрительного нерва и сужение сосудов сетчатки
Глазное дно типа «соль с перцем»: мелкие белые пятна и депигментация пигментного эпителия сетчатки
Костные тельца и субретинальные друзы (мраморное глазное дно)
Макулярная дегенерация и экссудация по типу болезни Коатса
На ОКТ наружные слои сетчатки почти полностью отсутствуют, характерно истончение или исчезновение эллипсоидной зоны (граница IS/OS). Данные различаются в зависимости от генотипа:
QДля чего нужен глазо-пальцевой признак (потирание глаз)?
A
Это характерное поведение пациентов с LCA: они трут или надавливают на глаза пальцами, чтобы механически стимулировать сетчатку и попытаться вызвать зрительное ощущение. Длительное повторение приводит к атрофии орбитальной жировой клетчатки и может вызвать энофтальм.
LCA — это группа наследственных дегенеративных заболеваний сетчатки, большинство из которых наследуются по аутосомно-рецессивному типу2). Редко мутации в CRX, IMPDH1 или OTX2 могут проявлять аутосомно-доминантное наследование2). Также сообщалось о X-сцепленном наследовании.
В настоящее время описано 19 типов LCA (LCA1–LCA19), а также 8 дополнительных ассоциированных генов2). Причинные гены вовлечены в несколько путей развития и функции сетчатки, включая морфогенез фоторецепторов, реснички фототрансдукции и зрительный цикл.
Гены, ассоциированные с LCA, классифицируются по пяти основным функциональным сетям2):
Метаболизм ретиноидов и палочковый зрительный цикл (RPE65, LRAT, RDH12)
Обнаружение световых стимулов и зрительное восприятие (GUCY2D, CNGA3)
Соединительные реснички фоторецепторов и поддержание наружного сегмента (CEP290, RPGRIP1, RPGR)
Наиболее часто мутирующие гены в мире и их доли следующие:
Ген
Доля
Вовлеченный путь
CEP290
Около 15%
Функция ресничек
GUCY2D
Около 12%
Передача светового сигнала (синтез цГМФ)
CRB1
Около 10%
Поддержание полярности клеток
RPE65
около 8%
Метаболизм ретиноидов
В японской когорте (34 семьи) анализ NGS показал частоту выявления около 56%, причем наиболее часто мутировавшими генами были CRB1, NMNAT1 и RPGRIP11).
Идентификация причинного гена напрямую связана с определением пригодности к лечению. В частности, подтверждение мутации RPE65 необходимо для оценки пригодности к генной терапии (воретиген непарвовек)2).
AIPL1 функционирует как специфический шаперон фосфодиэстеразы 6 (PDE6), которая отвечает за расщепление цГМФ в световой сигнальной трансдукции. Дефицит AIPL1 приводит к резкому снижению количества белка PDE6, что вызывает нарушение метаболизма цГМФ, дегенерацию фоторецепторов и раннюю слепоту2). Мутации AIPL1 составляют около 5–10% всех случаев LCA2).
QКакова вероятность того, что следующий ребенок унаследует LCA?
A
При аутосомно-рецессивном наследовании, если оба родителя являются носителями, вероятность того, что следующий ребенок будет поражен, составляет 25%, вероятность быть носителем — 50%, а вероятность быть здоровым и не носителем — 25%. Если причинный ген известен, возможна пренатальная или преимплантационная диагностика.
Диагноз LCA ставится клинически и требует подтверждения с помощью электроретинографии и молекулярно-генетического подтверждения с помощью генетического тестирования2).
LCA подозревается при врожденном тяжелом нарушении зрительной реакции (отсутствие фиксации и слежения). У младенцев с тяжелым нарушением зрения и высокой гиперметропией поиск LCA с помощью молекулярно-генетического тестирования является первым выбором4).
Основные диагностические исследования приведены ниже.
Исследование
Результаты / Особенности
Электроретинография (ЭРГ)
Не регистрируется или значительно снижена в темновых и световых условиях. Обязательно.
Различается в зависимости от подтипа (при типе RPE65 исчезает, при типе GUCY2D нормальная).
Генетическое тестирование (NGS и др.)
Необходимо для окончательного диагноза и идентификации подтипа.
Электроретинография: Ответы палочек и колбочек отсутствуют или значительно снижены. Нормальная ЭРГ исключает диагноз LCA.
ОКТ: Наружные слои сетчатки и эллипсоидная зона почти полностью исчезают. При мутациях CRB1 наблюдается парадоксальное утолщение сетчатки (грубая ламинация)1). Портативная ОКТ полезна для обследования бодрствующих младенцев или маленьких детей под наркозом4).
ФАФ: Результаты различаются в зависимости от подтипа. При мутациях GUCY2D аутофлуоресценция сохраняется нормальной, при мутациях RPE65 исчезает2).
Генетическое тестирование: Используются секвенирование нового поколения (NGS), ДНК-микрочипы, анализ сцепления и др. Общая частота выявления составляет около 70–80%2). С 2023 года панельное тестирование на 82 гена, вызывающих IRD (панель PrismGuide IRD), покрывается страховкой и применяется у молодых пациентов с подозрением на RPE65-ассоциированную IRD.
Для большинства форм LCA не установлено эффективного лечения. Текущее ведение следующее:
Коррекция аномалий рефракции : Соответствующая коррекция сильной гиперметропии и т.д. Может быть сильная аномалия рефракции, поэтому выписать очки и поощрять ортоптическое лечение.
Ортоптическое лечение : Визуальная реабилитация для максимального использования остаточной зрительной функции.
Обучение использованию вспомогательных средств : Из-за значительного нарушения зрительной функции рассмотреть обучение шрифту Брайля, тренировку ходьбы с белой тростью, использование увеличительных устройств и т.д.
Помощь слабовидящим : Использование вспомогательных средств для слабовидящих, поддержка оптимального доступа к образованию и трудоустройству.
Генетическое консультирование : Рекомендуется для семей и пациентов. Возможно тестирование носительства, пренатальная диагностика, преимплантационная диагностика.
Борьба со светобоязнью : Рекомендуется использование солнцезащитных очков и уменьшение воздействия света.
Регулярное наблюдение : Офтальмологическое наблюдение, включая электроретинограмму, при необходимости направление к специалисту по слабовидению.
В 2017 году FDA США одобрило воретиген непарвовек-rzyl (торговое название Luxturna) для лечения LCA2, связанной с биаллельными мутациями RPE65. Это первый продукт генной терапии, одобренный FDA в офтальмологии. В 2023 году он был одобрен также в Японии (название продукта: Luxturna®).
Рекомбинантный аденоассоциированный вирусный вектор (rAAV2) используется для введения нормальной копии гена RPE65 в пигментный эпителий сетчатки путем субретинальной инъекции. Процедура проводится в операционной для витрэктомии.
Исследование фазы III 301 (Russell 2017)5):
Субъекты: 31 пациент с RPE65-ассоциированной IRD
Первичная конечная точка: MLMT (тест мобильности при различной освещенности)
Также отмечено значительное улучшение по порогу стимуляции полного поля (FST)
Долгосрочные результаты фазы I/III (Maguire 2019)6):
Чувствительность сетчатки, острота зрения и функциональные преимущества, достигающие пика через 6–12 месяцев после лечения, впоследствии демонстрировали тенденцию к прогрессирующему снижению.
Повышение чувствительности сетчатки может улучшить ночную слепоту и поле зрения.
Национальное исследование фазы III (A11301) (Fujinami 2025)7):
Субъекты: 4 японских пациента с RPE65-ассоциированной IRD
Значительное повышение чувствительности по FST (определяемое как повышение чувствительности более чем в 10 раз)
Расширение поля зрения подтверждено через 1 год после введения.
Основные нежелательные явления: глазные нарушения, включая боль в глазу (предположительно связанные с процедурой введения)
Протокол введения:
Второй глаз вводится не менее чем через 6 дней после первого
Иммуносупрессия: начало приема стероидов за 3 дня до введения, продолжение в течение 14 дней после
Однако мутации RPE65 составляют лишь около 8% всех пациентов с LCA. Для других типов мутаций в настоящее время не существует доказанных эффективных методов лечения.
QМожно ли использовать генную терапию для всех пациентов с LCA?
A
В настоящее время одобренная генная терапия (воретиген непарвовек / Луксурна®) показана только для LCA2, вызванной биаллельными мутациями RPE65. Мутации RPE65 составляют около 8% всех LCA, и большинство пациентов не имеют показаний. Лечение для других генотипов находится на стадии исследований.
QКак получить генную терапию в Японии?
A
Условиями являются подтверждение биаллельной мутации RPE65 и наличие достаточного количества жизнеспособных клеток сетчатки. Генетическая диагностика с помощью панельного теста (PrismGuide IRD panel) является первым шагом. Рекомендуется направление в специализированный центр, имеющий опыт лечения наследственных заболеваний сетчатки.
Патофизиология LCA связана с нарушением зрительного цикла (Visual Cycle), из-за чего глаз не может передавать световую информацию.
Зрительный цикл представляет собой серию ферментативных реакций между пигментным эпителием сетчатки (RPE) и нейросенсорной сетчаткой, которые метаболизируют пищевой витамин А для производства 11-цис-ретиналя и генерации зрительных пигментов. Без 11-цис-ретиналя каскад фототрансдукции не может начаться, и зрительные нервные сигналы не передаются в зрительную кору. Мутация в любом из генов, кодирующих белки, участвующие в этой цепи реакций, может блокировать зрительный цикл и вызывать симптомы LCA.
Гистопатологически показано вовлечение наружной сетчатки и фоторецепторов, что позволяет предположить, что LCA является дегенеративным процессом, а не дисплазией.
GUCY2D (LCA1) : кодирует специфичную для сетчатки гуанилатциклазу (GC-E). Катализирует синтез цГМФ, ключевой элемент фототрансдукции. Идентифицировано более 140 болезнь-ассоциированных мутаций, 88% являются причиной аутосомно-рецессивной LCA. Аминокислота 838 известна как горячая точка мутаций 2).
RPE65 (LCA2) : принадлежит к суперсемейству каротиноид-расщепляющих оксигеназ. Это бифункциональный фермент, катализирующий расщепление O-алкилового эфира из all-trans-ретинилового эфира и изомеризацию ретинильной части (all-trans-ретинол → 11-cis-ретинол) 2). Необходим для функции как палочек, так и колбочек. Недавние исследования предполагают возможное участие в изомеризации лютеина в мезо-зеаксантин 2). Единственное одобренное показание для генной терапии.
CRB1 (LCA8) : гомолог белка crumbs дрозофилы, экспрессируется во внутреннем сегменте фоторецепторов и клетках Мюллера. Важен для поддержания клеточной полярности, расположен на хромосоме 1q31.3 1).
CEP290 (LCA10) : участвует в функции ресничек фоторецепторов. Наиболее часто мутирующий ген среди генов, ассоциированных с LCA (около 15%).
NMNAT1 (LCA9) : кодирует ключевой фермент биосинтеза НАД (никотинамидадениндинуклеотида) 2).
LCA5 : кодирует леберцилин, участвующий в функции ресничек и внутриресничном транспорте белков 2).
AIPL1 : функционирует как специальный шаперон для PDE6 (фосфодиэстеразы 6). Дефицит AIPL1 дестабилизирует PDE6 → нарушение метаболизма цГМФ → аномалия каналов → дегенерация фоторецепторов 2).
7. Новейшие исследования и перспективы (отчеты на стадии исследований)
Долгосрочное наблюдение воретигена непарвовек (NCT00481546, NCT00643747) сообщает о начальном пике через 6–12 месяцев после лечения, за которым следует прогрессирующее снижение клинических преимуществ, включая чувствительность сетчатки, остроту зрения и функциональные улучшения 6). Исследование PERCEIVE (проспективное регистровое исследование) сообщило о 2-летних данных безопасности и эффективности в реальной клинической практике, при этом увеит, ассоциированный с генной терапией (GTAU), наблюдался в до 50% случаев 9).
Разработано Editas Medicine. Вектор AAV5 несет Cas9 из S. aureus и две направляющие РНК, нацеленные на глубокую интронную мутацию (c.2991+1655A>G) в интроне 26 гена CEP2903). Первое исследование на человеке подтвердило безопасность и хорошую переносимость даже при относительно высоких дозах3).
Восстановление зрительной функции с помощью оптогенетики
В качестве генно-независимого подхода изучается метод экспрессии светочувствительного каналородопсина в сохранившихся внутренних нейронах сетчатки3). Он может быть применим ко всем типам LCA (независимо от генотипа), и проводятся ранние клинические испытания.
Генная терапия для мутаций GUCY2D и AIPL1 проводится на животных моделях и показывает многообещающие результаты в спасении палочковых и колбочковых фоторецепторов.
Течение LCA классифицируется на три паттерна: стабильное (около 75%), прогрессирующее ухудшение (около 15%) и улучшение (около 10%). Мутации AIPL1 связаны с прогрессирующим ухудшением, тогда как мутации RPGRIP1 ассоциированы со стабильным течением. В будущем ожидается остановка прогрессирования или лечение с помощью искусственного зрения, генной терапии или регенеративной медицины.
Duan W, Zhou T, Jiang H, Zhang M, Hu M, Zhang L. A novel nonsense variant (c.1499C>G) in CRB1 caused Leber congenital amaurosis-8 in a Chinese family and a literature review. BMC Med Genomics. 2022;15(1):197. doi:10.1186/s12920-022-01356-z.
Mordà D, Alibrandi S, Scimone C, Rinaldi C, Scalinci SZ, Abate G, et al. Decoding pediatric inherited retinal dystrophies: Bridging genetic complexity and clinical heterogeneity. Progress in retinal and eye research. 2025;109:101405. doi:10.1016/j.preteyeres.2025.101405. PMID:40998214.
Botto C, Rucli M, Tekinsoy MD, Pulman J, Sahel JA, Dalkara D. Early and late stage gene therapy interventions for inherited retinal degenerations. Progress in retinal and eye research. 2022;86:100975. doi:10.1016/j.preteyeres.2021.100975. PMID:34058340.
Gurnani B, et al. Clinical approach to pediatric nystagmus: a comprehensive diagnostic algorithm. Clin Ophthalmol. 2025;19:1617-1636.
Russell S, Bennett J, Wellman JA, Chung DC, Yu ZF, Tillman A, et al. Efficacy and safety of voretigene neparvovec (AAV2-hRPE65v2) in patients with RPE65-mediated inherited retinal dystrophy: a randomised, controlled, open-label, phase 3 trial. Lancet (London, England). 2017;390(10097):849-860. doi:10.1016/S0140-6736(17)31868-8. PMID:28712537; PMCID:PMC5726391.
Maguire AM, Russell S, Wellman JA, Chung DC, Yu ZF, Tillman A, et al. Efficacy, Safety, and Durability of Voretigene Neparvovec-rzyl in RPE65 Mutation-Associated Inherited Retinal Dystrophy: Results of Phase 1 and 3 Trials. Ophthalmology. 2019;126(9):1273-1285. doi:10.1016/j.ophtha.2019.06.017. PMID:31443789.
Fujinami K, Akiyama K, Tsunoda K, et al. Efficacy and Safety of Voretigene Neparvovec in RPE65-Retinopathy: Results of a Phase III Trial in Japan. Ophthalmol Sci. 2025;5:100876. doi:10.1016/j.xops.2025.100876. PMID:40910102; PMCID:PMC12405627.
Gange WS, Sisk RA, Besirli CG, Lee TC, Havunjian M, Schwartz H, et al. Perifoveal Chorioretinal Atrophy after Subretinal Voretigene Neparvovec-rzyl for RPE65-Mediated Leber Congenital Amaurosis. Ophthalmology. Retina. 2022;6(1):58-64. doi:10.1016/j.oret.2021.03.016. PMID:33838313; PMCID:PMC8497635.
Fischer MD, Simonelli F, Sahni J, Holz FG, Maier R, Fasser C, et al. Real-World Safety and Effectiveness of Voretigene Neparvovec: Results up to 2 Years from the Prospective, Registry-Based PERCEIVE Study. Biomolecules. 2024;14(1). doi:10.3390/biom14010122. PMID:38254722; PMCID:PMC10813228.
Скопируйте текст статьи и вставьте его в выбранный ИИ-ассистент.
Статья скопирована в буфер обмена
Откройте ИИ-ассистент ниже и вставьте скопированный текст в чат.