La amaurosis congénita de Leber (LCA) es la forma más grave de distrofia retiniana congénita, causando discapacidad visual severa desde el nacimiento hasta la infancia. Es una causa principal de discapacidad visual infantil y se conoce como una causa representativa de discapacidad visual congénita. Las manifestaciones clínicas son diversas.
Fue reportada por primera vez en 1869 por el oftalmólogo alemán Theodor Karl Gustav von Leber (1840–1917). Nótese que la neuropatía óptica hereditaria de Leber (LHON), reportada por el mismo Leber en 1871, es una enfermedad mitocondrial que se desarrolla alrededor de los 20 años y es completamente diferente de la LCA. En 1957, la ausencia de ondas en el electrorretinograma (ERG) se confirmó como una característica común del diagnóstico de LCA, estableciendo el nombre de la enfermedad.
La prevalencia estimada al nacer es de 2 a 3 por cada 100,000 nacimientos (1/30,000 a 1/81,000)1). Algunos informes citan 1:80,000 a 1:200,000, mostrando variabilidad2). La LCA representa aproximadamente el 5% de todas las distrofias retinianas, y alrededor del 20% de los niños con discapacidad visual que asisten a escuelas para ciegos tienen LCA1). Actualmente, se han identificado alrededor de 27 genes asociados con LCA2), y se identifican genes causantes en aproximadamente el 70–80% de los casos2). El patrón de herencia es principalmente autosómico recesivo, pero también se han reportado herencia autosómica dominante y ligada al cromosoma X.
Q¿Cuándo se suele detectar la amaurosis congénita de Leber?
A
Los padres a menudo notan nistagmo o falta de fijación alrededor de las 6 semanas de edad3). Cuando se observa una respuesta visual severamente deficiente (incapacidad total para fijar o seguir), se sospecha LCA y se confirma mediante electrorretinograma.
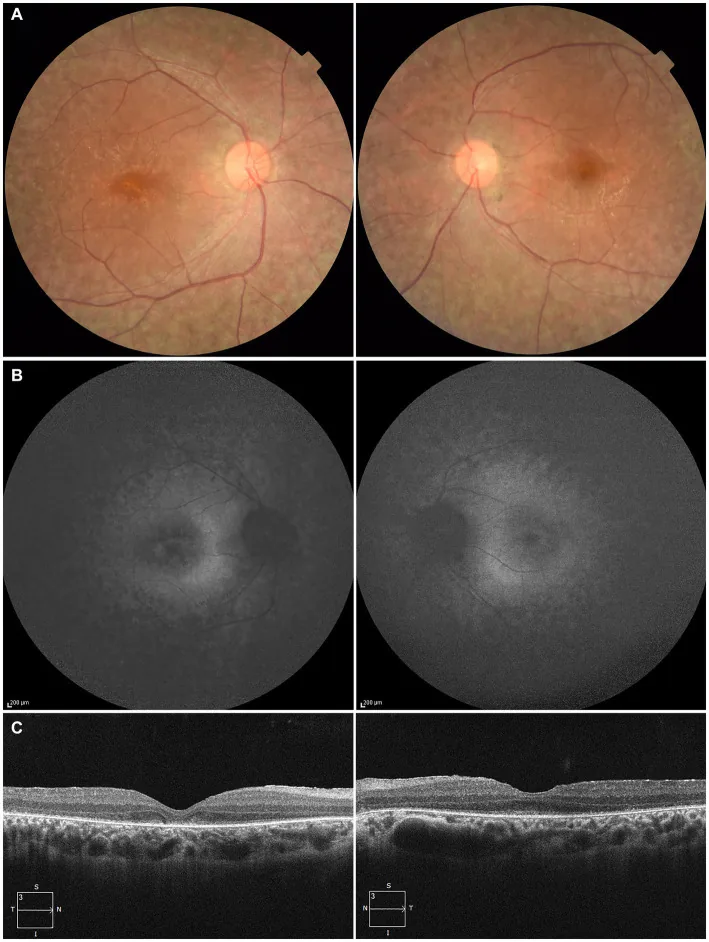
Higa N, et al. A novel RPE65 variant p.(Ala391Asp) in Leber congenital amaurosis: a case report and literature review in Japan. Front Med (Lausanne). 2024. Figure 1. PMCID: PMC11446184. License: CC BY.
Paciente de 27 años: (A) Fotografías de fondo de ojo de ambos ojos que muestran despigmentación del epitelio pigmentario de la retina y estrechamiento de los vasos retinianos; (B) Autofluorescencia de fondo que muestra hiperautofluorescencia y pérdida de señal en las áreas afectadas; (C) OCT que muestra pérdida de la zona elipsoide (EZ) en el ojo derecho y mínima EZ residual en la fóvea del ojo izquierdo. Corresponde a la degeneración del epitelio pigmentario de la retina y el estrechamiento de los vasos retinianos discutidos en la sección “2. Síntomas principales y hallazgos clínicos”.
Existe discapacidad visual grave desde el nacimiento o la primera infancia.
Pérdida de agudeza visual: La mayoría de los pacientes tienen una agudeza visual de 0.1 o menos, y aproximadamente un tercio no tiene percepción de luz. La discapacidad visual generalmente es estable o progresa muy lentamente.
Fotofobia: Muchos pacientes muestran sensibilidad a la luz.
Ceguera nocturna: La función visual en condiciones de oscuridad se reduce aún más.
Los padres a menudo notan nistagmo o falta de fijación alrededor de las 6 semanas de edad 3).
Hallazgos clínicos (hallazgos confirmados por el médico en el examen)
Nistagmo: Aparece al nacer o poco después. Es pendular o errante y está presente en todas las posiciones de la mirada. Este es un punto de diferenciación importante con la distrofia retiniana grave de inicio temprano (EOSRD), que no presenta nistagmo2).
Anomalía de la respuesta pupilar: El reflejo pupilar a la luz es lento o está ausente. Se denomina “pupilas amauróticas”.
Signo oculodigital: Acción de hurgar, presionar o frotar los ojos, que se cree que es un intento de estimular mecánicamente la retina para evocar sensaciones visuales. La principal secuela es la enoftalmia debida a la atrofia de la grasa orbitaria.
Error refractivo: La hipermetropía alta (>5 dioptrías) es común, y se cree que resulta de una alteración de la emetropización debido a la discapacidad visual temprana.
En la infancia, el fondo de ojo a menudo parece normal, pero posteriormente aparecen diversos hallazgos. Los hallazgos del fondo de ojo varían desde un fondo normal hasta un aspecto típico de retinitis pigmentosa. En casos avanzados, el disco óptico se vuelve pálido, los vasos sanguíneos se estrechan extremadamente, el color general del fondo se oscurece y el reflejo foveal desaparece.
Palidez del disco óptico y estrechamiento de los vasos retinianos
Fondo en sal y pimienta: degeneración del epitelio pigmentario de la retina con pequeñas manchas blancas y despigmentación
Pigmentación en espículas óseas y manchas subretinianas (fondo marmóreo)
En la OCT, las capas externas de la retina están casi ausentes, con adelgazamiento hasta pérdida de la zona elipsoide (unión IS/OS) como característica. Los hallazgos varían según el genotipo:
Mutación en CRB1: hallazgo paradójico de aumento del grosor retiniano (laminación gruesa)1)
Mutación en RPE65: ausencia de FAF (autofluorescencia del fondo de ojo)2)
Q¿Cuál es el propósito del signo oculodigital (frotarse los ojos)?
A
Es un comportamiento característico en pacientes con LCA, que se cree que estimula mecánicamente la retina al hurgar o presionar los ojos con los dedos, tratando de evocar sensaciones visuales. Con el tiempo, provoca atrofia de la grasa orbitaria y causa enoftalmos.
La LCA es un grupo de enfermedades degenerativas de la retina hereditarias, la mayoría con herencia autosómica recesiva2). Raramente, mutaciones en CRX, IMPDH1 u OTX2 muestran herencia autosómica dominante2). También se ha reportado herencia ligada al cromosoma X.
Actualmente se han reportado 19 tipos (LCA1 a LCA19) y 8 genes relacionados adicionales2). Los genes causantes están involucrados en múltiples vías relacionadas con el desarrollo y función de la retina, incluyendo la morfogénesis de los fotorreceptores, los cilios de fototransducción y el ciclo visual.
Los genes asociados a LCA se clasifican en cinco redes funcionales principales2):
Metabolismo de retinoides / ciclo visual de bastones (RPE65, LRAT, RDH12)
Mantenimiento de la homeostasis retiniana / mantenimiento de fotorreceptores (AIPL1, SPATA7, TULP1, USP45, CRB1, LCA5)
Desarrollo retiniano / morfogénesis (RD3, CEP290)
Detección de luz / percepción visual (GUCY2D, CNGA3)
Cilio conector del fotorreceptor / mantenimiento del segmento externo (CEP290, RPGRIP1, RPGR)
Los genes mutados más frecuentes a nivel mundial y sus proporciones son los siguientes:
Gen
Proporción
Vía implicada
CEP290
Aproximadamente 15%
Función ciliar
GUCY2D
Aproximadamente 12%
Fototransducción (síntesis de cGMP)
CRB1
Aproximadamente 10%
Mantenimiento de la polaridad celular
RPE65
Aproximadamente 8%
Metabolismo de retinoides
En una cohorte japonesa (34 familias), el análisis NGS mostró una tasa de detección de aproximadamente el 56%, y los genes mutados más frecuentes reportados fueron CRB1, NMNAT1 y RPGRIP11).
La identificación del gen causante determina directamente la elegibilidad para el tratamiento. En particular, la confirmación de la mutación RPE65 es esencial para determinar la elegibilidad para la terapia génica (voretigene neparvovec)2).
AIPL1 funciona como una chaperona específica de la fosfodiesterasa 6 (PDE6), que es responsable de la degradación de cGMP en la fototransducción. La deficiencia de AIPL1 conduce a una reducción drástica de la proteína PDE6, lo que resulta en una alteración del metabolismo de cGMP, degeneración de fotorreceptores y ceguera temprana2). Las mutaciones en AIPL1 representan aproximadamente el 5-10% de todos los casos de LCA2).
Q¿Cuál es la probabilidad de que el próximo hijo también tenga LCA?
A
En la herencia autosómica recesiva, si ambos padres son portadores, la probabilidad de que el próximo hijo esté afectado es del 25%, la probabilidad de ser portador es del 50% y la probabilidad de no estar afectado ni ser portador es del 25%. Si se identifica el gen causante, el diagnóstico prenatal o el diagnóstico genético preimplantacional son opciones.
El diagnóstico de LCA se realiza clínicamente, y se requiere confirmación mediante electrorretinografía para el diagnóstico definitivo y pruebas genéticas para la confirmación molecular genética2).
Se sospecha LCA cuando hay una falta congénita marcada de respuesta visual (falta de fijación y seguimiento). En lactantes con discapacidad visual grave e hipermetropía alta, la prueba genética molecular para LCA es la primera opción4).
Las principales pruebas diagnósticas se muestran a continuación.
Varía según el subtipo (ausente en tipo RPE65, normal en tipo GUCY2D).
Prueba genética (NGS, etc.)
Necesaria para el diagnóstico definitivo y la identificación del subtipo.
Electrorretinograma: Tanto las respuestas de bastones como de conos están ausentes o marcadamente reducidas. Un ERG normal descarta LCA.
OCT: Las capas externas de la retina y la zona elipsoide están casi ausentes. Las mutaciones en CRB1 muestran un engrosamiento retiniano paradójico (laminación gruesa) 1). La OCT portátil es útil para examinar lactantes despiertos o niños pequeños bajo anestesia 4).
FAF: Los hallazgos varían según el subtipo. Las mutaciones en GUCY2D muestran autofluorescencia preservada, mientras que las mutaciones en RPE65 muestran autofluorescencia ausente 2).
Prueba genética: Se utilizan secuenciación de nueva generación (NGS), micromatrices de ADN y análisis de ligamiento. La tasa de detección general es de aproximadamente 70–80% 2). Desde 2023, una prueba de panel para 82 genes causantes de IRD (Panel PrismGuide IRD) está cubierta por el seguro y se aplica a pacientes de inicio joven con sospecha de IRD relacionada con RPE65.
Para la mayoría de los tipos de LCA, no se ha establecido un tratamiento sustancial. El manejo actual es el siguiente.
Corrección de errores refractivos: Proporcione la corrección refractiva adecuada para la hipermetropía alta, etc. Dado que pueden presentarse errores refractivos fuertes, prescriba gafas y realice entrenamiento visual.
Entrenamiento visual: Realice rehabilitación visual para maximizar el uso de la función visual residual.
Entrenamiento de sustitución visual: Debido a que la función visual está gravemente afectada, considere la instrucción en Braille, el entrenamiento para caminar con bastón blanco, el uso de lupas de lectura, etc.
Atención de baja visión: Apoye el uso de ayudas para baja visión y el acceso óptimo a oportunidades educativas y laborales.
Consejo genético: Recomendado para familias y pacientes. Pueden ser posibles las pruebas de portador, el diagnóstico prenatal y el diagnóstico genético preimplantacional.
Manejo de la fotofobia: Se recomienda el uso de gafas tintadas y la reducción de la exposición a la luz.
Seguimiento regular: Realice un seguimiento oftalmológico que incluya electrorretinografía y derive a una clínica de baja visión según sea necesario.
En 2017, la FDA de EE. UU. aprobó voretigene neparvovec-rzyl (nombre comercial Luxturna) como tratamiento para LCA2 asociado con mutaciones bialélicas en RPE65. Este es el primer producto de terapia génica aprobado por la FDA en el campo de la oftalmología. En 2023, también fue aprobado en Japón (nombre del producto: inyección Luxturna®).
Un vector de virus adenoasociado recombinante (rAAV2) introduce una copia normal del gen RPE65 en el epitelio pigmentario de la retina mediante inyección subretiniana. El procedimiento se realiza en un quirófano de cirugía vítrea.
Ensayo de fase III 301 (Russell 2017)5):
Sujetos: 31 pacientes con IRD asociada a RPE65
Criterio de valoración principal: MLMT (prueba de movilidad con múltiples luminancias; prueba de observación del comportamiento bajo diferentes niveles de iluminación)
También se observó una mejora significativa en el umbral de estímulo de campo completo (FST)
Resultados a largo plazo de fase I/III (Maguire 2019)6):
La sensibilidad retiniana, la agudeza visual y las ganancias funcionales que alcanzaron su punto máximo entre 6 y 12 meses después del tratamiento mostraron una tendencia a disminuir progresivamente después
Se puede esperar una mejora en la ceguera nocturna y el campo visual debido al aumento de la sensibilidad retiniana
Ensayo nacional de fase III (ensayo A11301) (Fujinami 2025)7):
Sujetos: 4 pacientes japoneses con IRD asociada a RPE65
Aumento significativo de la sensibilidad en FST (umbral de estímulo de campo completo) (definido como un aumento de sensibilidad de más de 10 veces)
Se confirmó la expansión del campo visual al año de la administración
Principales eventos adversos: trastornos oculares, incluido dolor ocular (presuntamente relacionados con el procedimiento de administración)
Protocolo de administración:
El segundo ojo se administra al menos 6 días después del primer ojo.
Inmunosupresión: iniciar esteroides 3 días antes de la administración y continuar durante 14 días después de la administración.
Sin embargo, las mutaciones en RPE65 representan solo aproximadamente el 8% de todos los pacientes con LCA. Para otros tipos de mutaciones, actualmente no existe un tratamiento probado.
Q¿Se puede usar la terapia génica en todos los pacientes con LCA?
A
Actualmente, la terapia génica aprobada (voretigene neparvovec / inyección Luxturna®) está indicada solo para LCA2 causada por mutaciones bialélicas en RPE65. Las mutaciones en RPE65 representan aproximadamente el 8% de todos los casos de LCA, por lo que la mayoría de los pacientes no son elegibles. Los tratamientos para otros genotipos aún se encuentran en etapa de investigación.
Q¿Cómo puedo recibir terapia génica en Japón?
A
Se requiere la confirmación de mutaciones bialélicas en RPE65 y suficientes células retinianas viables. El diagnóstico genético mediante una prueba de panel genético (panel PrismGuide IRD) es el primer paso. Se recomienda la derivación a un centro especializado con experiencia en el tratamiento de enfermedades retinianas hereditarias.
La fisiopatología de la LCA está relacionada con la interrupción del ciclo visual, que impide que el ojo transmita información lumínica.
El ciclo visual es una serie de reacciones enzimáticas entre el epitelio pigmentario de la retina (EPR) y la retina neurosensorial, que metaboliza la vitamina A de la dieta para producir 11-cis retinal, que genera fotopigmentos. Sin 11-cis retinal, la cascada de fototransducción no puede iniciarse y las señales nerviosas visuales no se transmiten a la corteza visual. Las mutaciones en cualquiera de los genes que codifican proteínas involucradas en esta cascada pueden bloquear el ciclo visual y causar síntomas de LCA.
Histopatológicamente, se ha demostrado la participación de la retina externa y los fotorreceptores, lo que sugiere que la LCA es un proceso degenerativo más que una malformación.
GUCY2D (LCA1): Codifica la guanilato ciclasa específica de la retina (GC-E). Cataliza la síntesis de cGMP y es clave en la fototransducción. Se han identificado más de 140 mutaciones asociadas a la enfermedad, el 88% causan LCA autosómica recesiva. El aminoácido 838 es un punto caliente de mutación conocido 2).
RPE65 (LCA2): Pertenece a la superfamilia de oxigenasas de escisión de carotenoides. Es una enzima bifuncional que cataliza la escisión del éster O-alquilo del éster all-trans-retinilo y la isomerización de la porción retinilo (all-trans-retinol → 11-cis-retinol) 2). Es esencial tanto para la función de bastones como de conos, y estudios recientes sugieren que también podría estar involucrada en la isomerización de luteína a meso-zeaxantina 2). Es la única indicación aprobada para terapia génica.
CRB1 (LCA8): Homólogo de la proteína crumbs de Drosophila, se expresa en los segmentos internos de los fotorreceptores y en las células de Müller. Importante para mantener la polaridad celular, ubicado en el cromosoma 1q31.3 1).
CEP290 (LCA10): Involucrado en la función ciliar de los fotorreceptores. Tiene la frecuencia de mutación más alta entre los genes asociados a LCA (aproximadamente 15%).
NMNAT1 (LCA9): Codifica una enzima clave en la biosíntesis de NAD (nicotinamida adenina dinucleótido) 2).
LCA5: Codifica lebercilina, involucrada en la función ciliar y el transporte intraflagelar 2).
AIPL1: Funciona como un chaperón especializado de PDE6 (fosfodiesterasa 6). La deficiencia de AIPL1 conduce a inestabilidad de PDE6 → alteración del metabolismo de cGMP → anomalías en los canales → degeneración de fotorreceptores 2).
7. Investigación más reciente y perspectivas futuras (informes en etapa de investigación)
Las evaluaciones de seguimiento a largo plazo de voretigene neparvovec (NCT00481546, NCT00643747) han informado que después de un pico inicial a los 6-12 meses posteriores al tratamiento, los beneficios clínicos que incluyen sensibilidad retiniana, agudeza visual y ganancias funcionales disminuyen progresivamente 6). El estudio PERCEIVE (estudio de registro prospectivo) informó datos de seguridad y eficacia a 2 años en la práctica clínica real, con GTAU (uveítis asociada a terapia génica) observada en hasta el 50% de los casos 9).
Desarrollado por Editas Medicine. Un vector AAV5 que transporta Cas9 derivado de S. aureus y dos ARN guía se dirige a la mutación intrónica profunda (c.2991+1655A>G) en el intrón 26 de CEP290 3). Se confirmó la seguridad en un primer ensayo en humanos, mostrando buena tolerabilidad incluso a dosis relativamente altas 3).
Optogenética para la restauración de la función visual
Como enfoque independiente del gen, se está investigando un método para expresar canalrodopsina sensible a la luz en las neuronas retinianas internas restantes 3). Puede ser aplicable a todos los tipos de LCA (independientemente del genotipo), y se están realizando ensayos clínicos iniciales.
La terapia génica para mutaciones en GUCY2D y AIPL1 está progresando en modelos animales, mostrando resultados prometedores en el rescate de fotorreceptores de bastones y conos.
El curso de la LCA se clasifica en tres patrones: estable (aproximadamente 75%), empeoramiento progresivo (aproximadamente 15%) y mejoría (aproximadamente 10%). Las mutaciones en AIPL1 se asocian con empeoramiento progresivo, mientras que las mutaciones en RPGRIP1 se asocian con un curso estable. En el futuro, se espera detener la progresión o tratar mediante visión artificial, terapia génica y medicina regenerativa.
Duan W, Zhou T, Jiang H, Zhang M, Hu M, Zhang L. A novel nonsense variant (c.1499C>G) in CRB1 caused Leber congenital amaurosis-8 in a Chinese family and a literature review. BMC Med Genomics. 2022;15(1):197. doi:10.1186/s12920-022-01356-z.
Mordà D, Alibrandi S, Scimone C, Rinaldi C, Scalinci SZ, Abate G, et al. Decoding pediatric inherited retinal dystrophies: Bridging genetic complexity and clinical heterogeneity. Progress in retinal and eye research. 2025;109:101405. doi:10.1016/j.preteyeres.2025.101405. PMID:40998214.
Botto C, Rucli M, Tekinsoy MD, Pulman J, Sahel JA, Dalkara D. Early and late stage gene therapy interventions for inherited retinal degenerations. Progress in retinal and eye research. 2022;86:100975. doi:10.1016/j.preteyeres.2021.100975. PMID:34058340.
Gurnani B, et al. Clinical approach to pediatric nystagmus: a comprehensive diagnostic algorithm. Clin Ophthalmol. 2025;19:1617-1636.
Russell S, Bennett J, Wellman JA, Chung DC, Yu ZF, Tillman A, et al. Efficacy and safety of voretigene neparvovec (AAV2-hRPE65v2) in patients with RPE65-mediated inherited retinal dystrophy: a randomised, controlled, open-label, phase 3 trial. Lancet (London, England). 2017;390(10097):849-860. doi:10.1016/S0140-6736(17)31868-8. PMID:28712537; PMCID:PMC5726391.
Maguire AM, Russell S, Wellman JA, Chung DC, Yu ZF, Tillman A, et al. Efficacy, Safety, and Durability of Voretigene Neparvovec-rzyl in RPE65 Mutation-Associated Inherited Retinal Dystrophy: Results of Phase 1 and 3 Trials. Ophthalmology. 2019;126(9):1273-1285. doi:10.1016/j.ophtha.2019.06.017. PMID:31443789.
Fujinami K, Akiyama K, Tsunoda K, et al. Efficacy and Safety of Voretigene Neparvovec in RPE65-Retinopathy: Results of a Phase III Trial in Japan. Ophthalmol Sci. 2025;5:100876. doi:10.1016/j.xops.2025.100876. PMID:40910102; PMCID:PMC12405627.
Gange WS, Sisk RA, Besirli CG, Lee TC, Havunjian M, Schwartz H, et al. Perifoveal Chorioretinal Atrophy after Subretinal Voretigene Neparvovec-rzyl for RPE65-Mediated Leber Congenital Amaurosis. Ophthalmology. Retina. 2022;6(1):58-64. doi:10.1016/j.oret.2021.03.016. PMID:33838313; PMCID:PMC8497635.
Fischer MD, Simonelli F, Sahni J, Holz FG, Maier R, Fasser C, et al. Real-World Safety and Effectiveness of Voretigene Neparvovec: Results up to 2 Years from the Prospective, Registry-Based PERCEIVE Study. Biomolecules. 2024;14(1). doi:10.3390/biom14010122. PMID:38254722; PMCID:PMC10813228.
Copia el texto del artículo y pégalo en el asistente de IA que prefieras.
Artículo copiado al portapapeles
Abre un asistente de IA abajo y pega el texto copiado en el chat.