Die Lebersche kongenitale Amaurose (LCA) ist die schwerste Form der angeborenen Netzhautdystrophie, die ab der Geburt oder im Säuglingsalter zu einer schweren Sehbehinderung führt. Sie ist eine der Hauptursachen für Sehbehinderung im Kindesalter und gilt als repräsentative Ursache für angeborene Sehbehinderung. Das klinische Bild ist vielfältig.
Sie wurde erstmals 1869 von dem deutschen Augenarzt Theodor Karl Gustav von Leber (1840–1917) beschrieben. Die ebenfalls von Leber 1871 beschriebene Lebersche hereditäre Optikusneuropathie (LHON) ist eine Mitochondrienerkrankung, die etwa im Alter von 20 Jahren auftritt und sich völlig von der LCA unterscheidet. 1957 wurde das Fehlen von Wellen im Elektroretinogramm (ERG) als gemeinsames Merkmal der LCA-Diagnose bestätigt, und der Krankheitsname etablierte sich.
Die geschätzte Geburtsprävalenz liegt bei 2 bis 3 pro 100.000 Geburten (1/30.000 bis 1/81.000)1). Einige Berichte geben 1:80.000 bis 1:200.000 an, was auf eine Schwankungsbreite hinweist2). Sie macht etwa 5 % aller Netzhautdystrophien aus, und etwa 20 % der sehbehinderten Kinder, die Blindenschulen besuchen, haben eine LCA1). Derzeit sind etwa 27 LCA-assoziierte Gene identifiziert2), und bei etwa 70–80 % der Fälle wird das ursächliche Gen gefunden2). Der Vererbungsmodus ist hauptsächlich autosomal-rezessiv, es gibt jedoch auch Berichte über dominante und X-chromosomale Vererbung.
QWann wird die Lebersche kongenitale Amaurose normalerweise erkannt?
A
In der Regel bemerken die Eltern im Alter von etwa 6 Wochen einen Nystagmus oder fehlende Fixation3). Bei schwerer Sehreaktionsstörung (keinerlei Fixation oder Verfolgung) wird eine LCA vermutet und durch Elektroretinogramm bestätigt.
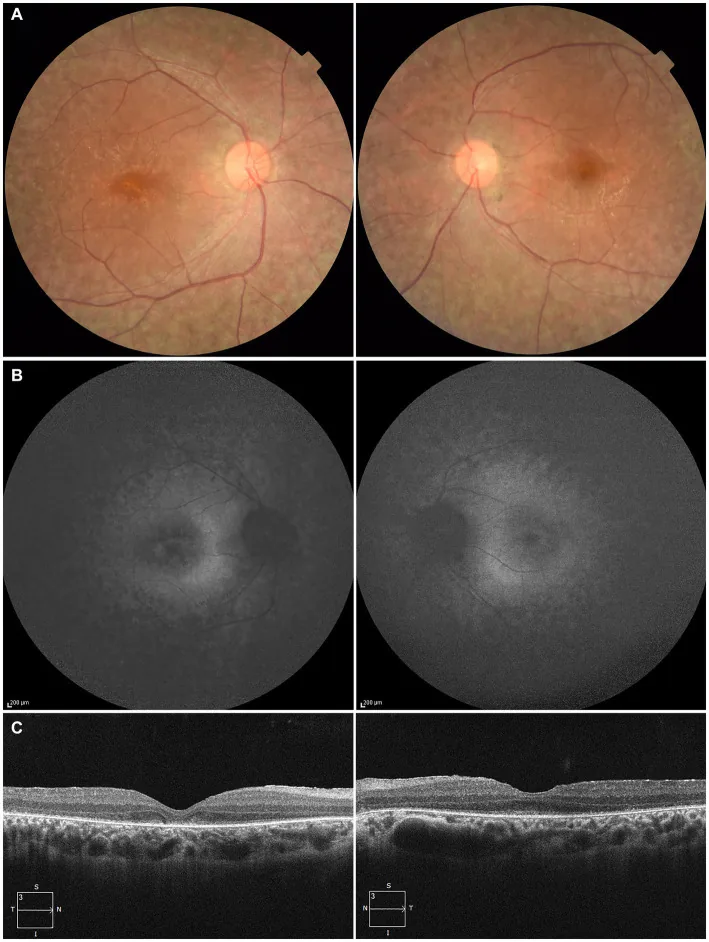
Higa N, et al. A novel RPE65 variant p.(Ala391Asp) in Leber congenital amaurosis: a case report and literature review in Japan. Front Med (Lausanne). 2024. Figure 1. PMCID: PMC11446184. License: CC BY.
Bei einem 27-jährigen Patienten zeigen (A) Fundusfotografien beider Augen eine Depigmentierung des retinalen Pigmentepithels und eine Verengung der Netzhautgefäße, (B) Fundusautofluoreszenz eine Hyperfluoreszenz und Signalabnahme in den Läsionsbereichen, (C) OCT eine Aufhebung der Ellipsoidzone (EZ) im rechten Auge und einen geringen Rest in der Fovea des linken Auges. Dies entspricht der Degeneration des retinalen Pigmentepithels und der Verengung der Netzhautgefäße, die im Abschnitt „2. Hauptsymptome und klinische Befunde“ behandelt werden.
Eine schwere Sehbehinderung besteht von Geburt an oder kurz nach der Geburt.
Verminderte Sehschärfe: Die meisten Patienten haben eine Sehschärfe unter 0,1, und etwa ein Drittel hat keine Lichtwahrnehmung. Die Sehbehinderung ist in der Regel stabil oder schreitet sehr langsam fort.
Photophobie: Viele Patienten zeigen eine Überempfindlichkeit gegenüber Licht.
Nachtblindheit: Die Sehfunktion bei Dunkelheit ist noch weiter reduziert.
Eltern bemerken oft im Alter von etwa 6 Wochen einen Nystagmus oder fehlende Fixation 3).
Klinische Befunde (vom Arzt bei der Untersuchung festgestellt)
Nystagmus: Tritt bei der Geburt oder kurz danach auf. Er ist pendelnd oder wandernd und in allen Blickrichtungen vorhanden. Dies ist ein wichtiges Unterscheidungsmerkmal zur Early-Onset Severe Retinal Dystrophy (EOSRD), die ohne Nystagmus einhergeht 2).
Pupillenreaktionsstörung: Der Lichtreflex ist träge bis aufgehoben. Man spricht von „amaurotischen Pupillen“.
Oculo-digitales Zeichen: Augen werden gestoßen, gedrückt oder gerieben, was als mechanischer Stimulationsversuch der Netzhaut zur Hervorrufung von Seheindrücken gedeutet wird. Hauptfolge ist eine Enophthalmus durch orbitale Fettatrophie.
Refraktionsfehler: Eine hohe Hyperopie (>5 Dioptrien) ist häufig, vermutlich aufgrund einer Störung der Emmetropisation durch die frühe Sehbehinderung.
Im Säuglingsalter kann der Fundus oft normal erscheinen, später treten jedoch vielfältige Befunde auf. Die Fundusbefunde reichen von normalem Fundus bis zu einem typischen Retinitis-pigmentosa-ähnlichen Fundus. In fortgeschrittenen Fällen wird die Papille blass, die Gefäße werden stark verengt, die Gesamtfarbe des Fundus wird dunkel und der Makularingreflex verschwindet.
Blässe der Papille und Verengung der Netzhautgefäße
Salz-und-Pfeffer-Fundus: kleine weiße Flecken und Depigmentierung des retinalen Pigmentepithels
Knochenbälkchenartige Pigmentierung und subretinale Drusen (marmorierter Fundus)
Im OCT sind die äußeren Netzhautschichten fast vollständig verschwunden, mit einer Ausdünnung bis zum Verlust der Ellipsoidzone (IS/OS-Grenze). Die Befunde variieren je nach Genotyp:
CRB1-Mutation: paradoxe Verdickung der Netzhaut (grobe Schichtung) 1)
QWozu dient das okulo-digitale Zeichen (Augenreiben)?
A
Es ist ein charakteristisches Verhalten von LCA-Patienten: Sie reiben oder drücken mit den Fingern auf die Augen, um die Netzhaut mechanisch zu stimulieren und so eine visuelle Wahrnehmung hervorzurufen. Langfristige Wiederholung führt zur Atrophie des orbitalen Fettgewebes und kann eine Enophthalmie verursachen.
LCA ist eine Gruppe erblicher Netzhautdegenerationserkrankungen, die meist autosomal-rezessiv vererbt werden2). Selten können Mutationen in CRX, IMPDH1 oder OTX2 einen autosomal-dominanten Erbgang zeigen2). Auch X-chromosomale Vererbung wurde berichtet.
Derzeit sind 19 Typen (LCA1 bis LCA19) sowie 8 weitere assoziierte Gene beschrieben2). Die ursächlichen Gene sind an mehreren Wegen der Netzhautentwicklung und -funktion beteiligt, darunter Photorezeptor-Morphogenese, Phototransduktionszilien und der Sehzyklus.
LCA-assoziierte Gene werden in fünf Hauptfunktionsnetzwerke eingeteilt2):
Retinoidstoffwechsel und Stäbchen-Sehzyklus (RPE65, LRAT, RDH12)
Aufrechterhaltung der Netzhaut-Homöostase und der Photorezeptoren (AIPL1, SPATA7, TULP1, USP45, CRB1, LCA5)
Netzhautentwicklung und Morphogenese (RD3, CEP290)
Lichtreizdetektion und visuelle Wahrnehmung (GUCY2D, CNGA3)
Verbindungszilien der Photorezeptoren und Erhalt des Außensegments (CEP290, RPGRIP1, RPGR)
Die weltweit häufigsten mutierten Gene und ihre Anteile sind wie folgt:
Gen
Anteil
Beteiligter Signalweg
CEP290
Etwa 15 %
Zilienfunktion
GUCY2D
Etwa 12 %
Lichtsignalübertragung (cGMP-Synthese)
CRB1
Etwa 10 %
Aufrechterhaltung der Zellpolarität
RPE65
etwa 8 %
Retinoidstoffwechsel
In einer japanischen Kohorte (34 Familien) zeigte die NGS-Analyse eine Nachweisrate von etwa 56 %, wobei die häufigsten mutierten Gene CRB1, NMNAT1 und RPGRIP1 waren1).
Die Identifizierung des ursächlichen Gens steht in direktem Zusammenhang mit der Bestimmung der Behandlungseignung. Insbesondere der Nachweis einer RPE65-Mutation ist für die Beurteilung der Eignung für eine Gentherapie (Voretigen Neparvovec) unerlässlich2).
AIPL1 fungiert als spezifisches Chaperon für Phosphodiesterase 6 (PDE6), das in der Lichtsignaltransduktion für den Abbau von cGMP verantwortlich ist. Ein AIPL1-Mangel führt zu einer drastischen Abnahme der PDE6-Proteinmenge, was zu einer Störung des cGMP-Stoffwechsels, einer Degeneration der Photorezeptoren und früher Erblindung führt2). AIPL1-Mutationen machen etwa 5–10 % aller LCA-Fälle aus2).
QWie hoch ist die Wahrscheinlichkeit, dass das nächste Kind LCA erbt?
A
Bei autosomal-rezessiver Vererbung beträgt die Wahrscheinlichkeit, dass das nächste Kind betroffen ist, 25 %, Träger zu sein 50 % und weder betroffen noch Träger zu sein 25 %, wenn beide Eltern Träger sind. Wenn das ursächliche Gen bekannt ist, sind Pränataldiagnostik oder Präimplantationsdiagnostik möglich.
Die Diagnose der LCA erfolgt klinisch und erfordert eine Bestätigung durch Elektroretinographie und eine molekulargenetische Bestätigung durch Gentests2).
Bei angeborener schwerer Sehreaktion (fehlende Fixation und Verfolgung) wird LCA vermutet. Bei Säuglingen mit schwerer Sehbehinderung und hoher Hypermetropie ist die Suche nach LCA mittels molekulargenetischer Tests die erste Wahl4).
Die wichtigsten diagnostischen Untersuchungen sind unten aufgeführt.
Unterschiedlich je nach Subtyp (bei RPE65-Typ verschwunden, bei GUCY2D-Typ normal).
Gentest (NGS usw.)
Notwendig für die endgültige Diagnose und Subtypidentifikation.
Elektroretinogramm: Sowohl die Stäbchen- als auch die Zapfenantwort fehlen oder sind deutlich vermindert. Ein normales ERG schließt die Diagnose einer LCA aus.
OCT: Die äußeren Netzhautschichten und die Ellipsoidzone sind nahezu verschwunden. Bei CRB1-Mutationen zeigt sich eine paradoxe Netzhautverdickung (grobe Lamellierung)1). Handgehaltene OCT-Geräte sind nützlich für die Untersuchung wacher Säuglinge oder kleiner Kinder unter Narkose4).
FAF: Die Befunde variieren je nach Subtyp. Bei GUCY2D-Mutationen bleibt die Autofluoreszenz normal erhalten, bei RPE65-Mutationen verschwindet sie2).
Gentest: Next-Generation Sequencing (NGS), DNA-Microarrays, Kopplungsanalyse usw. werden eingesetzt. Die Gesamtnachweisrate liegt bei etwa 70–80 %2). Seit 2023 ist ein Panel-Test für 82 IRD-verursachende Gene (PrismGuide IRD-Panel) von der Krankenkasse erstattungsfähig und wird bei jungen Patienten mit Verdacht auf RPE65-assoziierte IRD angewendet.
Für die meisten Formen der LCA ist keine kurative Behandlung etabliert. Das derzeitige Management ist wie folgt:
Korrektur von Refraktionsfehlern : Angemessene Korrektur von starker Hyperopie usw. Es kann eine starke Refraktionsstörung vorliegen, daher Brillenverordnung und Sehtraining fördern.
Sehtraining : Visuelle Rehabilitation zur maximalen Nutzung der verbleibenden Sehfunktion.
Training mit Sehhilfen : Aufgrund der erheblichen Beeinträchtigung der Sehfunktion sind Braille-Unterricht, Blindenstocktraining, Verwendung von Bildschirmlesegeräten usw. in Betracht zu ziehen.
Low-Vision-Versorgung : Verwendung von Low-Vision-Hilfsmitteln, Unterstützung beim optimalen Zugang zu Bildung und Beschäftigung.
Genetische Beratung : Für Familien und Patienten empfohlen. Trägertests, Pränataldiagnostik, Präimplantationsdiagnostik können möglich sein.
Umgang mit Photophobie : Verwendung von getönten Brillen und Reduzierung der Lichteinwirkung wird empfohlen.
Regelmäßige Nachsorge : Ophthalmologische Nachsorge einschließlich Elektroretinogramm, ggf. Überweisung an eine Low-Vision-Ambulanz.
Im Jahr 2017 genehmigte die US-amerikanische FDA Voretigen Neparvovec-rzyl (Handelsname Luxturna) zur Behandlung von LCA2 im Zusammenhang mit biallelischen RPE65-Mutationen. Es ist das erste von der FDA zugelassene Gentherapieprodukt in der Augenheilkunde. 2023 wurde es auch in Japan zugelassen (Produktname: Luxturna®).
Ein rekombinanter Adeno-assoziierter Virus (rAAV2)-Vektor wird verwendet, um eine normale Kopie des RPE65-Gens durch subretinale Injektion in das retinale Pigmentepithel einzubringen. Der Eingriff findet im Vitrektomie-Operationssaal statt.
Phase-III-301-Studie (Russell 2017)5):
Probanden: 31 Patienten mit RPE65-assoziierter IRD
Auch im Full-Field Stimulus Threshold (FST) wurde eine signifikante Verbesserung festgestellt.
Langzeitergebnisse der Phase I/III (Maguire 2019)6):
Die Netzhautempfindlichkeit, Sehschärfe und funktionellen Gewinne, die 6–12 Monate nach der Behandlung ihren Höhepunkt erreichten, zeigten anschließend eine tendenziell fortschreitende Abnahme.
Durch die Erhöhung der Netzhautempfindlichkeit können Nachtblindheit und Gesichtsfeld verbessert werden.
Nationale Phase-III-Studie (A11301) (Fujinami 2025)7):
Probanden: 4 japanische Patienten mit RPE65-assoziierter IRD
Signifikanter Anstieg der Empfindlichkeit im FST (definiert als mehr als 10-facher Anstieg der Empfindlichkeit)
Gesichtsfelderweiterung wurde 1 Jahr nach der Verabreichung bestätigt.
Wichtigste unerwünschte Ereignisse: Augenveränderungen einschließlich Augenschmerzen (vermutlich im Zusammenhang mit dem Verabreichungsverfahren)
Verabreichungsprotokoll:
Das zweite Auge wird mindestens 6 Tage nach dem ersten behandelt
Immunsuppression: Beginn der Steroide 3 Tage vor der Verabreichung, Fortsetzung für 14 Tage danach
Allerdings machen RPE65-Mutationen nur etwa 8 % aller LCA-Patienten aus. Für andere Mutationstypen gibt es derzeit keine nachweislich wirksame Behandlung.
QIst die Gentherapie für alle LCA-Patienten anwendbar?
A
Derzeit ist die zugelassene Gentherapie (Voretigen Neparvovec / Luxturna®) nur für LCA2 aufgrund biallelischer RPE65-Mutationen indiziert. RPE65-Mutationen machen etwa 8 % aller LCA aus, und die Mehrheit der Patienten hat keine Indikation. Behandlungen für andere Genotypen befinden sich in der Forschung.
QWie kann man in Japan eine Gentherapie erhalten?
A
Voraussetzungen sind der Nachweis einer biallelischen RPE65-Mutation und ausreichend lebensfähige Netzhautzellen. Die genetische Diagnose mittels Gen-Panel-Test (PrismGuide IRD-Panel) ist der erste Schritt. Eine Überweisung an ein spezialisiertes Zentrum mit Erfahrung in der Behandlung erblicher Netzhauterkrankungen wird empfohlen.
6. Pathophysiologie und detaillierter Krankheitsmechanismus
Die Pathophysiologie der LCA hängt mit der Störung des Sehzyklus (Visual Cycle) zusammen, wodurch das Auge keine Lichtinformationen übertragen kann.
Der Sehzyklus ist eine Reihe von enzymatischen Reaktionen zwischen dem retinalen Pigmentepithel (RPE) und der neurosensorischen Netzhaut, die Vitamin A aus der Nahrung verstoffwechselt, um 11-cis-Retinal zu produzieren und Sehpigmente zu erzeugen. Ohne 11-cis-Retinal kann die Phototransduktionskaskade nicht beginnen, und visuelle Nervensignale werden nicht an den visuellen Kortex weitergeleitet. Eine Mutation in einem der Gene, die für Proteine in dieser Reaktionskette kodieren, kann den Sehzyklus blockieren und die Symptome der LCA verursachen.
Histopathologisch wurde eine Beteiligung der äußeren Netzhaut und der Photorezeptoren nachgewiesen, was darauf hindeutet, dass es sich bei der LCA eher um einen degenerativen Prozess als um eine Dysplasie handelt.
GUCY2D (LCA1) : Kodiert für die netzhautspezifische Guanylatcyclase (GC-E). Katalysiert die cGMP-Synthese, Schlüssel zur Phototransduktion. Über 140 krankheitsassoziierte Mutationen identifiziert, 88% sind Ursache der autosomal-rezessiven LCA. Aminosäure 838 ist als Mutations-Hotspot bekannt 2).
RPE65 (LCA2) : Gehört zur Superfamilie der Carotinoid-spaltenden Oxygenasen. Es ist ein bifunktionelles Enzym, das die O-Alkylester-Spaltung von all-trans-Retinylester und die Isomerisierung des Retinylanteils (all-trans-Retinol → 11-cis-Retinol) katalysiert 2). Essentiell für die Funktion von Stäbchen und Zapfen. Neuere Studien deuten auf eine mögliche Beteiligung an der Isomerisierung von Lutein zu meso-Zeaxanthin hin 2). Einzige zugelassene Indikation für Gentherapie.
CRB1 (LCA8) : Homolog zum Drosophila-Crumbs-Protein, exprimiert im inneren Segment der Photorezeptoren und in Müller-Zellen. Wichtig für die Aufrechterhaltung der Zellpolarität, lokalisiert auf Chromosom 1q31.3 1).
CEP290 (LCA10) : Beteiligt an der Zilienfunktion der Photorezeptoren. Das am häufigsten mutierte Gen unter den LCA-assoziierten Genen (ca. 15%).
NMNAT1 (LCA9) : Kodiert für ein Schlüsselenzym der NAD-Biosynthese (Nicotinamidadenindinukleotid) 2).
LCA5 : Kodiert für Lebercilin, beteiligt an der Zilienfunktion und dem intraziliären Proteintransport 2).
AIPL1 : Fungiert als spezielles Chaperon für PDE6 (Phosphodiesterase 6). AIPL1-Mangel destabilisiert PDE6 → cGMP-Stoffwechselstörung → Kanalanomalie → Photorezeptordegeneration 2).
7. Aktuelle Forschung und Zukunftsaussichten (Forschungsstadium)
Die Langzeit-Nachbeobachtung von Voretigen Neparvovec (NCT00481546, NCT00643747) berichtet einen anfänglichen Peak 6–12 Monate nach der Behandlung, gefolgt von einer fortschreitenden Abnahme der klinischen Vorteile, einschließlich Netzhautempfindlichkeit, Sehschärfe und funktioneller Gewinne 6). Die PERCEIVE-Studie (prospektive Registerstudie) berichtete über 2-Jahres-Sicherheits- und Wirksamkeitsdaten aus der klinischen Praxis, wobei in bis zu 50% der Fälle eine Gentherapie-assoziierte Uveitis (GTAU) beobachtet wurde 9).
Entwickelt von Editas Medicine. Der AAV5-Vektor trägt Cas9 von S. aureus und zwei Guide-RNAs, die auf die tiefe Intron-Mutation (c.2991+1655A>G) im Intron 26 von CEP290 abzielen3). Die First-in-Human-Studie bestätigte die Sicherheit und zeigte eine gute Verträglichkeit auch bei relativ hohen Dosen3).
Wiederherstellung der Sehfunktion durch Optogenetik
Als genunabhängiger Ansatz wird eine Methode untersucht, bei der lichtempfindliches Channelrhodopsin in verbleibenden inneren Netzhautneuronen exprimiert wird3). Sie könnte auf alle LCA-Typen anwendbar sein (unabhängig vom Genotyp), und frühe klinische Studien laufen.
Die Gentherapie für GUCY2D- und AIPL1-Mutationen läuft in Tiermodellen und zeigt vielversprechende Ergebnisse bei der Rettung von Stäbchen- und Zapfenphotorezeptoren.
Der Verlauf der LCA wird in drei Muster eingeteilt: stabil (ca. 75%), fortschreitende Verschlechterung (ca. 15%) und Besserung (ca. 10%). AIPL1-Mutationen sind mit fortschreitender Verschlechterung verbunden, während RPGRIP1-Mutationen mit einem stabilen Verlauf assoziiert sind. Zukünftig werden ein Stopp des Fortschreitens oder eine Behandlung durch künstliches Sehen, Gentherapie oder regenerative Medizin erwartet.
Duan W, Zhou T, Jiang H, Zhang M, Hu M, Zhang L. A novel nonsense variant (c.1499C>G) in CRB1 caused Leber congenital amaurosis-8 in a Chinese family and a literature review. BMC Med Genomics. 2022;15(1):197. doi:10.1186/s12920-022-01356-z.
Mordà D, Alibrandi S, Scimone C, Rinaldi C, Scalinci SZ, Abate G, et al. Decoding pediatric inherited retinal dystrophies: Bridging genetic complexity and clinical heterogeneity. Progress in retinal and eye research. 2025;109:101405. doi:10.1016/j.preteyeres.2025.101405. PMID:40998214.
Botto C, Rucli M, Tekinsoy MD, Pulman J, Sahel JA, Dalkara D. Early and late stage gene therapy interventions for inherited retinal degenerations. Progress in retinal and eye research. 2022;86:100975. doi:10.1016/j.preteyeres.2021.100975. PMID:34058340.
Gurnani B, et al. Clinical approach to pediatric nystagmus: a comprehensive diagnostic algorithm. Clin Ophthalmol. 2025;19:1617-1636.
Russell S, Bennett J, Wellman JA, Chung DC, Yu ZF, Tillman A, et al. Efficacy and safety of voretigene neparvovec (AAV2-hRPE65v2) in patients with RPE65-mediated inherited retinal dystrophy: a randomised, controlled, open-label, phase 3 trial. Lancet (London, England). 2017;390(10097):849-860. doi:10.1016/S0140-6736(17)31868-8. PMID:28712537; PMCID:PMC5726391.
Maguire AM, Russell S, Wellman JA, Chung DC, Yu ZF, Tillman A, et al. Efficacy, Safety, and Durability of Voretigene Neparvovec-rzyl in RPE65 Mutation-Associated Inherited Retinal Dystrophy: Results of Phase 1 and 3 Trials. Ophthalmology. 2019;126(9):1273-1285. doi:10.1016/j.ophtha.2019.06.017. PMID:31443789.
Fujinami K, Akiyama K, Tsunoda K, et al. Efficacy and Safety of Voretigene Neparvovec in RPE65-Retinopathy: Results of a Phase III Trial in Japan. Ophthalmol Sci. 2025;5:100876. doi:10.1016/j.xops.2025.100876. PMID:40910102; PMCID:PMC12405627.
Gange WS, Sisk RA, Besirli CG, Lee TC, Havunjian M, Schwartz H, et al. Perifoveal Chorioretinal Atrophy after Subretinal Voretigene Neparvovec-rzyl for RPE65-Mediated Leber Congenital Amaurosis. Ophthalmology. Retina. 2022;6(1):58-64. doi:10.1016/j.oret.2021.03.016. PMID:33838313; PMCID:PMC8497635.
Fischer MD, Simonelli F, Sahni J, Holz FG, Maier R, Fasser C, et al. Real-World Safety and Effectiveness of Voretigene Neparvovec: Results up to 2 Years from the Prospective, Registry-Based PERCEIVE Study. Biomolecules. 2024;14(1). doi:10.3390/biom14010122. PMID:38254722; PMCID:PMC10813228.
Kopieren Sie den Artikeltext und fügen Sie ihn in den KI-Assistenten Ihrer Wahl ein.
Artikel in die Zwischenablage kopiert
Öffnen Sie unten einen KI-Assistenten und fügen Sie den kopierten Text in den Chat ein.