آموروز مادرزادی لبر (Leber congenital amaurosis; LCA) شدیدترین نوع دیستروفی شبکیه مادرزادی است که از بدو تولد تا شیرخوارگی باعث اختلال شدید بینایی میشود. این بیماری یکی از علل اصلی نابینایی در کودکان و نماینده علل مادرزادی نابینایی است. تظاهرات بالینی متنوع هستند.
این بیماری اولین بار در سال ۱۸۶۹ توسط چشمپزشک آلمانی تئودور فون لبر (Theodor Karl Gustav von Leber, 1840–1917) گزارش شد. توجه داشته باشید که نوروپاتی ارثی بینایی لبر (LHON) که توسط همان لبر در سال ۱۸۷۱ گزارش شد، یک بیماری میتوکندریایی با شروع حدود ۲۰ سالگی است و کاملاً با LCA متفاوت است. در سال ۱۹۵۷، فقدان امواج در الکترورتینوگرافی (ERG) به عنوان ویژگی مشترک تشخیص LCA تأیید شد و نام بیماری تثبیت گردید.
شیوع تخمینی هنگام تولد ۲ تا ۳ نفر در هر ۱۰۰٬۰۰۰ تولد (۱/۳۰٬۰۰۰ تا ۱/۸۱٬۰۰۰) است1). برخی منابع ۱:۸۰٬۰۰۰ تا ۱:۲۰۰٬۰۰۰ را ذکر کردهاند که نشاندهنده دامنه وسیع است2). LCA حدود ۵٪ از کل دیستروفیهای شبکیه را تشکیل میدهد و حدود ۲۰٪ از کودکان کمبینا که در مدارس نابینایان تحصیل میکنند، مبتلا به LCA هستند1). تاکنون حدود ۲۷ ژن مرتبط با LCA شناسایی شده است2) و در حدود ۷۰ تا ۸۰٪ موارد، ژن عامل مشخص میشود2). الگوی توارث عمدتاً اتوزومال مغلوب است، اما موارد اتوزومال غالب و وابسته به X نیز گزارش شده است.
Qآموروز مادرزادی لبر معمولاً چه زمانی تشخیص داده میشود؟
A
معمولاً والدین در حدود ۶ هفتگی به دلیل نیستاگموس یا عدم تثبیت نگاه متوجه مشکل میشوند3). در صورت مشاهده واکنش بینایی شدیداً ضعیف (عدم تثبیت و تعقیب)، LCA مشکوک شده و با الکترورتینوگرافی تشخیص قطعی داده میشود.
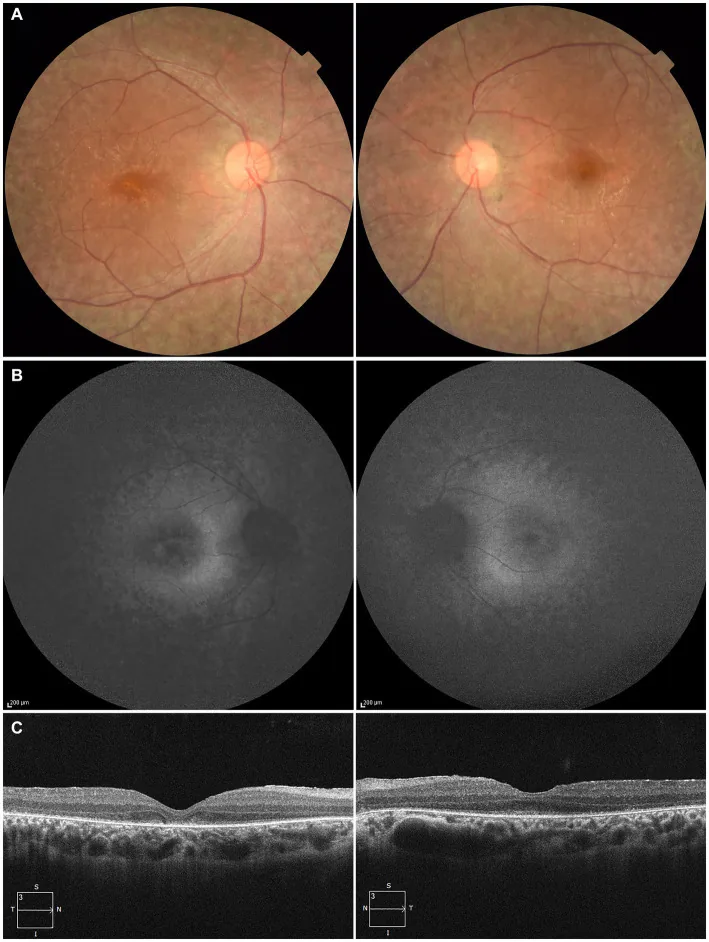
Higa N, et al. A novel RPE65 variant p.(Ala391Asp) in Leber congenital amaurosis: a case report and literature review in Japan. Front Med (Lausanne). 2024. Figure 1. PMCID: PMC11446184. License: CC BY.
در یک بیمار ۲۷ ساله، (A) عکس فوندوس دو چشم: دپیگمانتاسیون اپیتلیوم رنگدانه شبکیه و باریک شدن عروق شبکیه، (B) خودفلورسانس فوندوس: هیپرفلورسانس و کاهش سیگنال در نواحی ضایعه، (C) OCT: از بین رفتن ناحیه بیضوی (EZ) در چشم راست و باقیماندن اندک آن در فووئا چشم چپ. این یافتهها با دژنراسیون اپیتلیوم رنگدانه شبکیه و باریک شدن عروق شبکیه که در بخش «۲. علائم اصلی و یافتههای بالینی» بحث شده است، مطابقت دارد.
از بدو تولد یا اوایل پس از تولد، اختلال شدید بینایی وجود دارد.
کاهش بینایی: اکثر بیماران دارای دید کمتر از ۰.۱ هستند و حدود یک سوم هیچ درک نوری ندارند. اختلال بینایی معمولاً پایدار است یا بسیار آهسته پیشرفت میکند.
فوتوفوبی (نورگریزی): بسیاری از بیماران به نور حساسیت نشان میدهند.
شبکوری: عملکرد بینایی در محیط تاریک بیشتر کاهش مییابد.
اغلب والدین در حدود ۶ هفتگی به دلیل نیستاگموس یا عدم تثبیت نگاه متوجه مشکل میشوند3).
یافتههای بالینی (یافتههایی که پزشک در معاینه تأیید میکند)
نیستاگموس: از بدو تولد یا بلافاصله پس از آن ظاهر میشود. به صورت آونگی یا سرگردان در تمام جهات نگاه دیده میشود. این یک نکته افتراقی مهم با دیستروفی شدید شبکیه با شروع زودرس (EOSRD) است که در آن نیستاگموس وجود ندارد2).
واکنش غیرطبیعی مردمک: رفلکس نوری مردمک کند تا غایب است. به آن «مردک آموروتیک» گفته میشود.
علامت چشمی-دستی (oculo-digital sign): حرکات فرو کردن، فشار دادن یا مالیدن چشمها که تصور میشود تلاشی برای تحریک مکانیکی شبکیه و ایجاد حس بینایی است. عارضه اصلی آن فرورفتگی چشم به دلیل آتروفی چربی حدقه است.
عیوب انکساری: هایپروپی شدید (بیش از ۵ دیوپتر) شایع است که احتمالاً به دلیل عدم امتروپیزایی ناشی از اختلال بینایی اولیه است.
در دوران نوزادی، فوندوس اغلب طبیعی به نظر میرسد، اما بعداً یافتههای متنوعی ظاهر میشود. یافتههای فوندوس از فوندوس طبیعی تا فوندوس شبیه رتینیت پیگمانتوزا بسیار متنوع است. در موارد پیشرفته، دیسک بینایی رنگپریده، عروق بسیار باریک، رنگ کلی فوندوس تیره و رفلکس حلقوی ماکولا از بین میرود.
رنگپریدگی دیسک بینایی و باریک شدن عروق شبکیه
فوندوس نمک و فلفلی: دژنراسیون اپیتلیوم رنگدانه شبکیه با لکههای سفید کوچک و دپیگمانتاسیون
رسوب رنگدانه خارمانند و فلکهای زیرشبکیهای (فوندوس مرمری)
در OCT، لایههای خارجی شبکیه تقریباً به طور کامل از بین رفته و نازک شدن تا ناپدید شدن ناحیه بیضوی (اتصال IS/OS) مشخص است. یافتهها بر اساس ژنوتیپ متفاوت است:
Qعلامت چشمفشاری (مالیدن چشمها) برای چه انجام میشود؟
A
این رفتار مشخصه بیماران LCA است و تصور میشود که با فشار دادن یا مالیدن چشمها با انگشتان، شبکیه را به صورت مکانیکی تحریک کرده و سعی در برانگیختن بینایی دارند. تکرار طولانی مدت منجر به آتروفی چربی حدقه و فرورفتگی کره چشم میشود.
LCA گروهی از بیماریهای ارثی تحلیلبرنده شبکیه است که اکثراً به صورت اتوزومال مغلوب به ارث میرسند2). به ندرت، جهش در CRX، IMPDH1 و OTX2 باعث توارث اتوزومال غالب میشود2). مواردی از توارث وابسته به X نیز گزارش شده است.
در حال حاضر، علاوه بر 19 نوع LCA1 تا LCA19، هشت ژن مرتبط دیگر نیز گزارش شده است2). ژنهای عامل در مسیرهای متعددی از جمله مورفوژنز گیرنده نوری، مژک انتقال سیگنال نوری، و چرخه بینایی (چرخه دید) که در رشد و عملکرد شبکیه نقش دارند، دخیل هستند.
ژنهای مرتبط با LCA به پنج شبکه عملکردی اصلی طبقهبندی میشوند2):
متابولیسم رتینوئید و چرخه بینایی میلهای (RPE65, LRAT, RDH12)
حفظ هموستاز شبکیه و حفظ گیرندههای نوری (AIPL1, SPATA7, TULP1, USP45, CRB1, LCA5)
تکامل و مورفوژنز شبکیه (RD3, CEP290)
تشخیص تحریک نوری و ادراک بینایی (GUCY2D, CNGA3)
مژک اتصالی گیرنده نوری و حفظ بخش خارجی (CEP290, RPGRIP1, RPGR)
شایعترین ژنهای جهشیافته در جهان و درصد آنها به شرح زیر است:
ژن
درصد
مسیر درگیر
CEP290
حدود ۱۵٪
عملکرد مژک
GUCY2D
حدود ۱۲٪
انتقال پیام نوری (سنتز cGMP)
CRB1
حدود ۱۰٪
حفظ قطبیت سلولی
RPE65
حدود ۸٪
متابولیسم رتینوئید
در یک گروه ژاپنی (۳۴ خانواده)، تجزیه و تحلیل NGS میزان تشخیص را حدود ۵۶٪ نشان داد و شایعترین ژنهای جهشیافته CRB1، NMNAT1 و RPGRIP1 گزارش شدهاند1).
شناسایی ژن عامل به طور مستقیم با تعیین واجد شرایط بودن درمان مرتبط است. به ویژه، تأیید جهش RPE65 برای تعیین مناسب بودن ژن درمانی (voretigene neparvovec) ضروری است2).
AIPL1 به عنوان یک شپرون اختصاصی برای فسفودیاستراز ۶ (PDE6) عمل میکند که تجزیه cGMP را در انتقال پیام نوری بر عهده دارد. کمبود AIPL1 منجر به کاهش شدید پروتئین PDE6، اختلال در متابولیسم cGMP، تخریب گیرنده نوری و نابینایی زودرس میشود2). جهشهای AIPL1 حدود ۵ تا ۱۰٪ از کل موارد LCA را تشکیل میدهند2).
Qاحتمال انتقال LCA به فرزند بعدی چقدر است؟
A
در وراثت اتوزومال مغلوب، اگر هر دو والد ناقل باشند، احتمال ابتلای فرزند بعدی ۲۵٪، احتمال ناقلی ۵۰٪ و احتمال عدم ابتلا و عدم ناقلی ۲۵٪ است. در صورت شناسایی ژن عامل، تشخیص پیش از تولد یا تشخیص پیش از لانهگزینی نیز گزینههایی هستند.
تشخیص LCA به صورت بالینی انجام میشود و تأیید آن با الکترورتینوگرافی و تأیید مولکولی-ژنتیکی با آزمایش ژنتیک ضروری است2).
در صورت مشاهده پاسخ بینایی ضعیف مادرزادی (عدم تثبیت و دنبال کردن) باید به LCA مشکوک شد. در نوزادان با اختلال شدید بینایی و دوربینی بالا، جستجوی LCA با آزمایش ژنتیک مولکولی اولین انتخاب است4).
بسته به زیرگروه متفاوت است (در نوع RPE65 ناپدید میشود، در نوع GUCY2D طبیعی است)
آزمایش ژنتیکی (NGS و غیره)
برای تشخیص قطعی و تعیین زیرگروه ضروری است
الکترورتینوگرافی: پاسخهای استوانهای و مخروطی هر دو غیرقابل ثبت تا کاهش شدید هستند. الکترورتینوگرافی طبیعی تشخیص LCA را رد میکند.
OCT: لایههای خارجی شبکیه و ناحیه بیضوی تقریباً به طور کامل ناپدید میشوند. در جهشهای CRB1، افزایش متناقض ضخامت شبکیه (لایهبندی درشت) مشاهده میشود1). OCT دستی برای معاینه نوزادان بیدار یا کودکان زیر بیهوشی مفید است4).
FAF: یافتهها بسته به زیرگروه متفاوت است. در جهشهای GUCY2D فلورسانس خودبخودی طبیعی باقی میماند، در حالی که در جهشهای RPE65 ناپدید میشود2).
آزمایش ژنتیکی: از توالییابی نسل بعدی (NGS)، ریزآرایه DNA، آنالیز پیوستگی و غیره استفاده میشود. نرخ تشخیص کلی حدود 70-80٪ است2). از سال 2023، آزمایش پانل برای 82 ژن بیماریزای IRD (پانل PrismGuide IRD) تحت پوشش بیمه قرار گرفته و برای افراد جوان با مشکوک به IRD مرتبط با RPE65 اعمال میشود.
برای اکثر انواع LCA درمان مؤثری وجود ندارد. مدیریت فعلی به شرح زیر است:
اصلاح عیوب انکساری: اصلاح مناسب انکساری برای دوربینی شدید و غیره انجام میشود. ممکن است عیوب انکساری شدید وجود داشته باشد، بنابراین تجویز عینک و تلاش برای ارتوپتیک توصیه میشود.
ارتوپتیک: توانبخشی بینایی برای استفاده حداکثری از عملکرد بینایی باقیمانده انجام میشود.
آموزش جایگزین بینایی: به دلیل اختلال شدید بینایی، آموزش خط بریل، آموزش راه رفتن با عصای سفید و استفاده از ذرهبینهای بزرگکننده نیز در نظر گرفته میشود.
مراقبت از کمبینایان: استفاده از وسایل کمکبینایی و دسترسی بهینه به فرصتهای آموزشی و شغلی حمایت میشود.
مشاوره ژنتیک: برای خانواده و بیمار توصیه میشود. ممکن است آزمایش ناقل، تشخیص قبل از تولد و تشخیص قبل از لانهگزینی امکانپذیر باشد.
مدیریت فتوفوبی: استفاده از عینکهای محافظ نور و کاهش مواجهه با نور توصیه میشود.
پیگیری منظم: پیگیری چشمپزشکی شامل الکترورتینوگرام و در صورت لزوم ارجاع به کلینیک کمبینایان انجام میشود.
در سال ۲۰۱۷، سازمان غذا و داروی ایالات متحده (FDA) وورتیژن نپارووک (voretigene neparvovec-rzyl؛ با نام تجاری Luxturna) را به عنوان درمان LCA2 مرتبط با جهشهای دو آللی RPE65 تأیید کرد. این اولین محصول ژن درمانی تأیید شده توسط FDA در حوزه چشم پزشکی است. در سال ۲۰۲۳، این دارو در ژاپن نیز تأیید شد (نام محصول: Luxturna® تزریقی).
یک نسخه از ژن طبیعی RPE65 با استفاده از ناقل ویروس وابسته به آدنو نوترکیب (rAAV2) از طریق تزریق زیر شبکیه به اپیتلیوم رنگدانه شبکیه وارد میشود. این عمل در اتاق عمل جراحی ویتره انجام میشود.
کارآزمایی فاز III 301 (Russell 2017)5):
شرکتکنندگان: ۳۱ بیمار مبتلا به IRD مرتبط با RPE65
معیار پیامد اولیه: MLMT (آزمون تحرک با روشنایی چندگانه؛ یک آزمون مشاهده رفتاری در سطوح مختلف روشنایی)
آستانه محرک میدان کامل (FST) نیز بهبود معنیداری را نشان داد
نتایج بلندمدت فاز I/III (Maguire 2019)6):
حساسیت شبکیه، حدت بینایی و مزایای عملکردی که ۶ تا ۱۲ ماه پس از درمان به اوج خود رسیدند، تمایل به کاهش تدریجی در طول زمان نشان دادند
افزایش حساسیت شبکیه میتواند باعث بهبود شبکوری و میدان بینایی شود
کارآزمایی فاز III داخلی (آزمایش A11301) (Fujinami 2025)7):
شرکتکنندگان: ۴ بیمار ژاپنی مبتلا به IRD مرتبط با RPE65
افزایش معنیدار حساسیت در FST (آستانه محرک میدان کامل) (افزایش حساسیت بیش از ۱۰ برابر به عنوان معنیدار تعریف شد)
گسترش میدان بینایی یک سال پس از تجویز تأیید شد
عوارض جانبی اصلی: اختلالات چشمی از جمله درد چشم (که تصور میشود مربوط به روش تزریق باشد)
پروتکل تزریق:
دوز دوم حداقل ۶ روز پس از دوز اول تزریق میشود
سرکوب ایمنی: استروئید از ۳ روز قبل از تزریق شروع و تا ۱۴ روز پس از تزریق ادامه مییابد
با این حال، جهشهای RPE65 تنها حدود ۸٪ از کل بیماران LCA را تشکیل میدهند. برای سایر انواع جهش، در حال حاضر هیچ درمان اثبات شدهای وجود ندارد.
Qآیا ژن درمانی برای همه بیماران LCA قابل استفاده است؟
A
در حال حاضر، ژن درمانی تأیید شده (voretigene neparvovec / Luxturna®) فقط برای LCA2 ناشی از جهشهای دو آللی RPE65 قابل استفاده است. جهشهای RPE65 تنها حدود ۸٪ از کل موارد LCA را تشکیل میدهند و اکثر بیماران واجد شرایط نیستند. درمان برای سایر ژنوتیپها در مراحل تحقیقاتی است.
Qبرای دریافت ژن درمانی در ژاپن چه باید کرد؟
A
شرط لازم تأیید جهش دو آللی RPE65 و وجود سلولهای شبکیه زنده کافی است. اولین قدم تشخیص ژنتیکی با استفاده از پانل ژنی (PrismGuide IRD panel) است. ارجاع به مراکز تخصصی با تجربه در درمان بیماریهای ارثی شبکیه توصیه میشود.
پاتوفیزیولوژی LCA به اختلال در چرخه بینایی (Visual Cycle) مربوط میشود که در نتیجه چشم قادر به انتقال اطلاعات نوری نیست.
چرخه بینایی مجموعهای از واکنشهای آنزیمی بین اپیتلیوم رنگدانه شبکیه (RPE) و شبکیه حسی عصبی است که ویتامین A دریافتی از رژیم غذایی را متابولیزه کرده و ۱۱-سیس-رتینال (11-cis retinal) تولید میکند تا رنگدانه نوری را بسازد. بدون ۱۱-سیس-رتینال، آبشار انتقال اطلاعات نوری آغاز نمیشود و سیگنال عصبی بینایی به قشر بینایی منتقل نمیشود. جهش در هر یک از ژنهای کدکننده پروتئینهای دخیل در این زنجیره واکنشها میتواند چرخه بینایی را مسدود کرده و علائم LCA را ایجاد کند.
از نظر پاتولوژی بافتی، درگیری شبکیه خارجی و گیرندههای نوری نشان داده شده است که نشان میدهد LCA یک فرآیند دژنراتیو است نه یک ناهنجاری رشدی.
GUCY2D (LCA1): کدکننده گوانیلات سیکلاز اختصاصی شبکیه (GC-E). سنتز cGMP را کاتالیز کرده و کلید انتقال پیام نوری است. بیش از 140 جهش مرتبط با بیماری شناسایی شده که 88% آنها عامل LCA اتوزومال مغلوب هستند. اسید آمینه 838 به عنوان نقطه داغ جهش شناخته میشود 2).
RPE65 (LCA2): متعلق به ابرخانواده اکسیژنازهای برشدهنده کاروتنوئید است و آنزیمی دوکاره است که برش استر O-آلکیل از all-trans-retinyl ester و ایزومریزاسیون بخش رتینیل (all-trans-retinol → 11-cis-retinol) را کاتالیز میکند 2). برای عملکرد هر دو نوع سلول استوانهای و مخروطی ضروری است و اخیراً احتمال نقش آن در ایزومریزاسیون لوتئین به مزو-زآگزانتین مطرح شده است 2). تنها نشانه تأیید شده برای ژن درمانی.
CRB1 (LCA8): همولوگ با پروتئین crumbs در مگس سرکه، در بخش داخلی گیرنده نوری و سلولهای مولر بیان میشود. برای حفظ قطبیت سلولی مهم است و در کروموزوم 1q31.3 قرار دارد 1).
CEP290 (LCA10): در عملکرد مژک گیرندههای نوری نقش دارد. بیشترین فراوانی جهش را در میان ژنهای مرتبط با LCA دارد (حدود 15%).
NMNAT1 (LCA9): کدکننده آنزیم کلیدی در بیوسنتز NAD (نیکوتینآمید آدنین دینوکلئوتید) است 2).
LCA5: کدکننده لبرسیلین (lebercilin) که در عملکرد مژک و انتقال پروتئین درون مژکی نقش دارد 2).
AIPL1: به عنوان شپرون اختصاصی PDE6 (فسفودیاستراز 6) عمل میکند. کمبود AIPL1 منجر به ناپایداری PDE6 → اختلال در متابولیسم cGMP → ناهنجاری کانال → دژنراسیون گیرنده نوری میشود 2).
7. تحقیقات جدید و چشمانداز آینده (گزارشهای مرحله تحقیقاتی)
ارزیابی پیگیری بلندمدت وورتایژن نپارووک (NCT00481546، NCT00643747) نشان میدهد که پس از اوج اولیه در 6 تا 12 ماه پس از درمان، مزایای بالینی شامل حساسیت شبکیه، حدت بینایی و بهبود عملکرد به تدریج کاهش مییابد 6). مطالعه PERCEIVE (مطالعه ثبتنامی آیندهنگر) دادههای ایمنی و اثربخشی دو ساله را در عمل بالینی گزارش کرده است که در حداکثر 50% موارد یووئیت مرتبط با ژن درمانی (GTAU) مشاهده شده است 9).
توسعهیافته توسط Editas Medicine. این درمان از ناقل AAV5 حاوی Cas9 از S. aureus و دو RNA راهنما استفاده میکند و جهش اینترون عمیق (c.2991+1655A>G) در اینترون 26 ژن CEP290 را هدف قرار میدهد 3). در اولین مطالعه انسانی، ایمنی تأیید شد و نشان داده شد که حتی در دوزهای نسبتاً بالا نیز تحملپذیری خوبی دارد 3).
به عنوان یک رویکرد مستقل از ژن، روشی که در آن کانالرودوپسینهای حساس به نور در نورونهای باقیمانده شبکیه داخلی بیان میشود، در حال بررسی است 3). این روش به طور بالقوه برای همه انواع LCA (صرفنظر از ژنوتیپ) قابل استفاده است و آزمایشهای بالینی اولیه در حال انجام است.
ژن درمانی برای جهشهای GUCY2D و AIPL1 در مدلهای حیوانی در حال انجام است و نتایج امیدوارکنندهای در نجات گیرندههای نوری استوانهای و مخروطی نشان داده شده است.
سیر LCA به سه الگو تقسیم میشود: پایدار (حدود 75%)، بدتر شونده (حدود 15%) و بهبودی (حدود 10%). جهش AIPL1 با بدتر شدن پیشرونده و جهش RPGRIP1 با سیر پایدار مرتبط است. در آینده، توقف پیشرفت یا درمان با بینایی مصنوعی، ژن درمانی و پزشکی بازساختی نیز مورد انتظار است.
Duan W, Zhou T, Jiang H, Zhang M, Hu M, Zhang L. A novel nonsense variant (c.1499C>G) in CRB1 caused Leber congenital amaurosis-8 in a Chinese family and a literature review. BMC Med Genomics. 2022;15(1):197. doi:10.1186/s12920-022-01356-z.
Mordà D, Alibrandi S, Scimone C, Rinaldi C, Scalinci SZ, Abate G, et al. Decoding pediatric inherited retinal dystrophies: Bridging genetic complexity and clinical heterogeneity. Progress in retinal and eye research. 2025;109:101405. doi:10.1016/j.preteyeres.2025.101405. PMID:40998214.
Botto C, Rucli M, Tekinsoy MD, Pulman J, Sahel JA, Dalkara D. Early and late stage gene therapy interventions for inherited retinal degenerations. Progress in retinal and eye research. 2022;86:100975. doi:10.1016/j.preteyeres.2021.100975. PMID:34058340.
Gurnani B, et al. Clinical approach to pediatric nystagmus: a comprehensive diagnostic algorithm. Clin Ophthalmol. 2025;19:1617-1636.
Russell S, Bennett J, Wellman JA, Chung DC, Yu ZF, Tillman A, et al. Efficacy and safety of voretigene neparvovec (AAV2-hRPE65v2) in patients with RPE65-mediated inherited retinal dystrophy: a randomised, controlled, open-label, phase 3 trial. Lancet (London, England). 2017;390(10097):849-860. doi:10.1016/S0140-6736(17)31868-8. PMID:28712537; PMCID:PMC5726391.
Maguire AM, Russell S, Wellman JA, Chung DC, Yu ZF, Tillman A, et al. Efficacy, Safety, and Durability of Voretigene Neparvovec-rzyl in RPE65 Mutation-Associated Inherited Retinal Dystrophy: Results of Phase 1 and 3 Trials. Ophthalmology. 2019;126(9):1273-1285. doi:10.1016/j.ophtha.2019.06.017. PMID:31443789.
Fujinami K, Akiyama K, Tsunoda K, et al. Efficacy and Safety of Voretigene Neparvovec in RPE65-Retinopathy: Results of a Phase III Trial in Japan. Ophthalmol Sci. 2025;5:100876. doi:10.1016/j.xops.2025.100876. PMID:40910102; PMCID:PMC12405627.
Gange WS, Sisk RA, Besirli CG, Lee TC, Havunjian M, Schwartz H, et al. Perifoveal Chorioretinal Atrophy after Subretinal Voretigene Neparvovec-rzyl for RPE65-Mediated Leber Congenital Amaurosis. Ophthalmology. Retina. 2022;6(1):58-64. doi:10.1016/j.oret.2021.03.016. PMID:33838313; PMCID:PMC8497635.
Fischer MD, Simonelli F, Sahni J, Holz FG, Maier R, Fasser C, et al. Real-World Safety and Effectiveness of Voretigene Neparvovec: Results up to 2 Years from the Prospective, Registry-Based PERCEIVE Study. Biomolecules. 2024;14(1). doi:10.3390/biom14010122. PMID:38254722; PMCID:PMC10813228.
متن مقاله را کپی کنید و در دستیار هوش مصنوعی دلخواه خود بچسبانید.
مقاله در کلیپبورد کپی شد
یکی از دستیارهای هوش مصنوعی زیر را باز کنید و متن کپیشده را در کادر گفتگو بچسبانید.