الحثل الخلقي في شبكية العين من النوع ليبر (Leber congenital amaurosis; LCA) هو أشد أنواع الحثل الشبكي الخلقي، ويسبب ضعفًا بصريًا شديدًا منذ الولادة أو في مرحلة الرضاعة. وهو مرض رئيسي مسبب لضعف البصر لدى الأطفال، ويُعرف كسبب شائع للعمى الخلقي. تتنوع الصورة السريرية.
تم الإبلاغ عنه لأول مرة في عام 1869 من قبل طبيب العيون الألماني تيودور فون ليبر (Theodor Karl Gustav von Leber, 1840–1917). تجدر الإشارة إلى أن مرض الاعتلال العصبي البصري الوراثي ليبر (LHON) الذي أبلغ عنه ليبر نفسه في عام 1871 هو مرض ميتوكوندري يصيب في العقد الثاني من العمر، ويختلف تمامًا عن LCA. في عام 1957، تم تأكيد اختفاء الموجات في تخطيط كهربية الشبكية (ERG) كخاصية مشتركة لتشخيص LCA، مما أدى إلى ترسيخ اسم المرض.
يقدر معدل الانتشار عند الولادة بـ 2-3 لكل 100,000 مولود (1/30,000 إلى 1/81,000)1). بعض التقارير تشير إلى 1:80,000 إلى 1:200,000، مما يدل على تباين2). يشكل LCA حوالي 5% من جميع حالات الحثل الشبكي، وحوالي 20% من الأطفال ضعاف البصر في مدارس المكفوفين هم مصابون بـ LCA1). تم تحديد حوالي 27 جينًا مرتبطًا بـ LCA حتى الآن2)، ويتم تحديد الجين المسبب في حوالي 70-80% من الحالات2). النمط الوراثي السائد هو الوراثة الجسدية المتنحية، ولكن تم الإبلاغ عن حالات وراثة سائدة ومرتبطة بالكروموسوم X.
Qمتى يمكن اكتشاف الحثل الخلقي في شبكية العين من النوع ليبر؟
A
عادةً ما يلاحظ الوالدان الرأرأة أو عدم القدرة على التثبيت البصري في حوالي الأسبوع السادس من العمر3). في حالة وجود ضعف شديد في الاستجابة البصرية (عدم القدرة على التثبيت أو التتبع)، يُشتبه في LCA ويتم تأكيد التشخيص بواسطة تخطيط كهربية الشبكية.
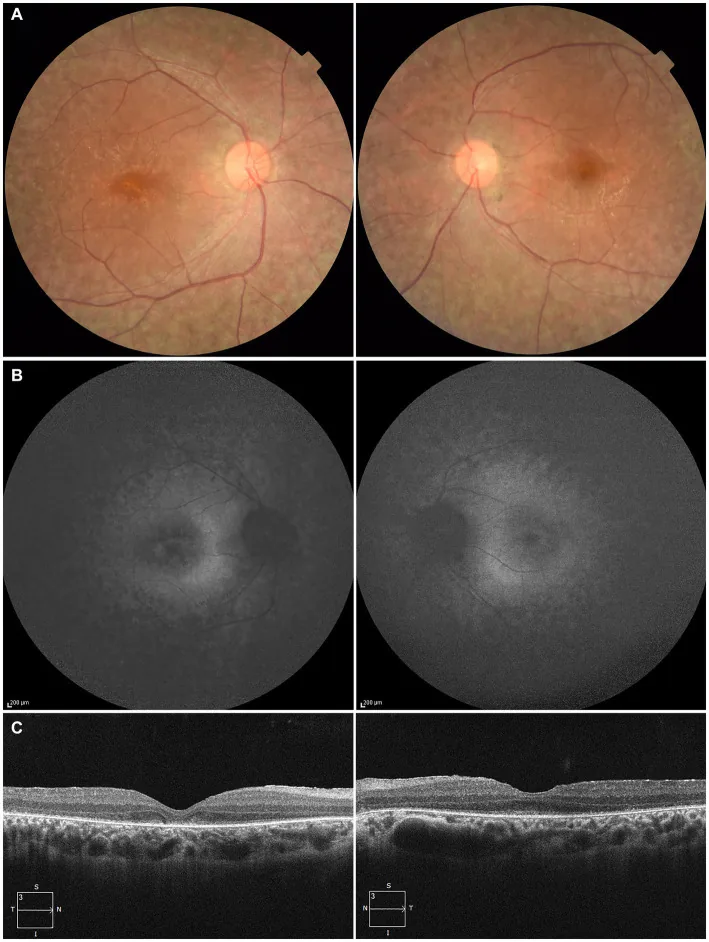
Higa N, et al. A novel RPE65 variant p.(Ala391Asp) in Leber congenital amaurosis: a case report and literature review in Japan. Front Med (Lausanne). 2024. Figure 1. PMCID: PMC11446184. License: CC BY.
لمريض يبلغ من العمر 27 عامًا، (أ) صورة قاع العين لكلا العينين تُظهر فقدان صبغة ظهارة الشبكية الصباغية وتضيق الأوعية الشبكية، (ب) التصوير الذاتي للقاع يُظهر فرط التألق وانخفاض الإشارة في المنطقة المصابة، (ج) التصوير المقطعي التوافقي البصري يُظهر اختفاء المنطقة الإهليلجية في العين اليمنى وبقاء ضئيل في النقرة اليسرى. يتوافق مع تنكس ظهارة الشبكية الصباغية وتضيق الأوعية الشبكية المذكورين في القسم “2. الأعراض الرئيسية والنتائج السريرية”.
الرأرأة: تظهر عند الولادة أو بعدها مباشرة. تكون بندولية أو عائمة في جميع اتجاهات النظر. وهي نقطة فارقة مهمة مع الحثل الشبكي الشديد المبكر (EOSRD)، حيث لا يصاحب EOSRD رأرأة2).
شذوذ رد فعل الحدقة: بطء أو اختفاء رد فعل الحدقة للضوء. يُسمى “حدقة عمياء (amaurotic pupils)”.
علامة العين والإصبع (oculo-digital sign): حركة دفع أو ضغط أو فرك العين، ويُعتقد أنها محاولة لتحفيز الشبكية ميكانيكيًا لإثارة الرؤية. المضاعفات الرئيسية هي غمور العين بسبب ضمور الدهون الحجاجية.
خطأ انكساري: طول النظر الشديد (>5 ديوبتر) شائع، ويُعتقد أنه ناتج عن ضعف التعادل البصري بسبب الضعف البصري المبكر.
في مرحلة الرضاعة، قد يبدو قاع العين طبيعيًا في كثير من الأحيان، ولكن تظهر نتائج متنوعة لاحقًا. تتنوع نتائج قاع العين من قاع طبيعي إلى قاع يشبه التهاب الشبكية الصباغي النمطي. في الحالات المتقدمة، يصبح القرص البصري شاحبًا، وتضيق الأوعية الدموية بشدة، ويصبح لون قاع العين العام داكنًا، ويختفي المنعكس الحلقي للبقعة.
شحوب القرص البصري وتضيق الأوعية الشبكية
قاع العين الملحي والفلفلي: تنكس ظهارة الشبكية الصباغية مع بقع بيضاء صغيرة ونقص التصبغ
تصبغات شوكية العظم واللطخات تحت الشبكية (قاع العين الرخامي)
في التصوير المقطعي التوافقي البصري (OCT)، تختفي الطبقات الخارجية للشبكية تقريبًا، ويتميز بترقق أو اختفاء المنطقة الإهليلجية (وصل IS/OS). تختلف النتائج حسب النمط الجيني:
طفرة CRB1: زيادة متناقضة في سمك الشبكية (تقسيم خشن) 1)
سلوك مميز لمرضى LCA، حيث يقومون بوخز أو الضغط على العين بأصابعهم لتحفيز الشبكية ميكانيكيًا ومحاولة استحضار الرؤية. يؤدي التكرار طويل الأمد إلى ضمور الدهون الحجاجية، مما يسبب غؤور العين.
LCA هي مجموعة من أمراض تنكس الشبكية الوراثية، ومعظمها يتبع نمط الوراثة الجسدية المتنحية2). نادرًا ما تظهر حالات الوراثة الجسدية السائدة بسبب طفرات في CRX وIMPDH1 وOTX22). كما توجد تقارير عن الوراثة المرتبطة بـ X.
حاليًا، تم الإبلاغ عن 19 نمطًا من LCA1 إلى LCA19، بالإضافة إلى 8 جينات مرتبطة أخرى2). تشارك الجينات المسببة في مسارات متعددة تتعلق بتطور ووظيفة الشبكية، مثل تشكل المستقبلات الضوئية، الأهداب الناقلة للضوء، والدورة البصرية.
تصنف جينات LCA المرتبطة إلى 5 شبكات وظيفية رئيسية2):
الأهداب المرتبطة بالمستقبلات الضوئية والحفاظ على القطعة الخارجية (CEP290, RPGRIP1, RPGR)
الجينات الأكثر شيوعًا للطفرات عالميًا ونسبها هي كما يلي:
الجين
النسبة
المسار المشارك
CEP290
حوالي 15%
وظيفة الأهداب
GUCY2D
حوالي 12%
نقل الإشارات الضوئية (تخليق cGMP)
CRB1
حوالي 10%
الحفاظ على الاستقطاب الخلوي
RPE65
حوالي 8%
أيض الريتينويد
في مجموعة يابانية (34 عائلة)، أظهر تحليل NGS معدل كشف حوالي 56%، وأكثر الجينات المتحورة شيوعًا هي CRB1 وNMNAT1 وRPGRIP11).
تحديد الجين المسبب يرتبط مباشرة بتحديد أهلية العلاج. على وجه الخصوص، تأكيد طفرة RPE65 ضروري لتقييم أهلية العلاج الجيني (voretigene neparvovec)2).
يعمل AIPL1 كمرافق خاص لإنزيم فوسفودايستراز 6 (PDE6)، الذي يقوم بتحلل cGMP في نقل الإشارات الضوئية. يؤدي نقص AIPL1 إلى انخفاض حاد في كمية بروتين PDE6، مما يؤدي إلى انهيار استقلاب cGMP → تنكس المستقبلات الضوئية → فقدان البصر المبكر2). تشكل طفرات AIPL1 حوالي 5-10% من جميع حالات LCA2).
Qما احتمال وراثة LCA للطفل التالي؟
A
في حالة الوراثة الجسدية المتنحية، إذا كان كلا الوالدين حاملين، فإن احتمال إصابة الطفل التالي هو 25%، واحتمال أن يكون حاملًا 50%، واحتمال عدم الإصابة وعدم الحمل 25%. إذا تم تحديد الجين المسبب، يمكن النظر في التشخيص قبل الولادة أو التشخيص قبل الزرع.
يتم تشخيص LCA سريريًا، ويتطلب تأكيدًا عن طريق تخطيط كهربية الشبكية وفحص جيني جزيئي2).
عند ملاحظة ضعف شديد في الاستجابة البصرية الخلقية (غياب التثبيت والتتبع)، يُشتبه في LCA. عند الرضع المصابين بضعف بصري شديد ومد بعيد شديد، يكون الفحص الجيني الجزيئي للبحث عن LCA هو الخيار الأول4).
OCT: الطبقات الخارجية للشبكية والمنطقة الإهليلجية مفقودة تقريبًا. في طفرات CRB1، يُلاحظ زيادة متناقضة في سمك الشبكية (تطبق خشن) 1). OCT المحمول مفيد لفحص الرضع المستيقظين والأطفال الصغار تحت التخدير 4).
FAF: تختلف النتائج حسب النوع الفرعي. في طفرات GUCY2D، يكون التألق الذاتي طبيعيًا؛ في طفرات RPE65، يختفي 2).
الاختبار الجيني: يُستخدم تسلسل الجيل التالي (NGS)، والمصفوفات الدقيقة للحمض النووي، وتحليل الارتباط. معدل الكشف الإجمالي حوالي 70-80% 2). منذ عام 2023، تم تغطية اختبار لوحة لـ 82 جينًا مسببًا لـ IRD (لوحة PrismGuide IRD) بالتأمين، ويُطبق على الحالات المبكرة المشتبه في إصابتها بـ IRD المرتبط بـ RPE65.
في عام 2017، وافقت إدارة الغذاء والدواء الأمريكية (FDA) على فوريتيجين نيبارفوفيك (voretigene neparvovec-rzyl؛ الاسم التجاري Luxturna) كعلاج لـ LCA2 المرتبط بطفرات RPE65 ثنائية الأليل. كان هذا أول منتج علاج جيني معتمد من إدارة الغذاء والدواء في مجال طب العيون. في عام 2023، تمت الموافقة عليه أيضًا في اليابان (اسم المنتج: Luxturna®注).
يتم إدخال نسخة طبيعية من جين RPE65 إلى الظهارة الصبغية للشبكية عن طريق الحقن تحت الشبكية باستخدام ناقل فيروس مرتبط بالغدة (rAAV2). يتم الإجراء في غرفة جراحة الجسم الزجاجي.
تجربة المرحلة الثالثة 301 (Russell 2017)5):
المشاركون: 31 مريضًا مصابًا بـ IRD مرتبط بـ RPE65
المقياس الأساسي: MLMT (اختبار التنقل متعدد الإضاءة؛ اختبار مراقبة السلوك في ظروف إضاءة مختلفة)
كما لوحظ تحسن كبير في عتبة التحفيز الكامل المجال (FST)
النتائج طويلة المدى للمرحلة الأولى/الثالثة (Maguire 2019)6):
أظهرت حساسية الشبكية وحدة البصر والمكاسب الوظيفية التي بلغت ذروتها بعد 6-12 شهرًا من العلاج ميلًا إلى الانخفاض التدريجي بعد ذلك
يمكن توقع تحسن العشى الليلي والمجال البصري نتيجة لزيادة حساسية الشبكية
التجربة المحلية من المرحلة الثالثة (تجربة A11301) (Fujinami 2025)7):
المشاركون: 4 مرضى يابانيين مصابين بـ IRD مرتبط بـ RPE65
زيادة كبيرة في الحساسية في FST (عتبة التحفيز الكامل المجال) (تم تعريف الزيادة في الحساسية بأكثر من 10 أضعاف على أنها كبيرة)
تم تأكيد توسع المجال البصري بعد عام واحد من العلاج
الآثار الضارة الرئيسية: اضطرابات العين بما في ذلك ألم العين (يفترض أنها مرتبطة بإجراء الحقن)
بروتوكول الإعطاء:
يتم إعطاء العين الثانية بعد 6 أيام على الأقل من العين الأولى
كبت المناعة: يبدأ الستيرويد قبل 3 أيام من الإعطاء ويستمر لمدة 14 يومًا بعد الإعطاء
ومع ذلك، فإن طفرات RPE65 تمثل حوالي 8% فقط من جميع مرضى LCA. لا يوجد علاج مثبت الفعالية حاليًا للأنماط الطفرية الأخرى.
Qهل يمكن استخدام العلاج الجيني لجميع مرضى LCA؟
A
العلاج الجيني المعتمد حاليًا (voretigene neparvovec / Luxturna®) مخصص فقط لمرض LCA2 الناتج عن طفرات ثنائية الأليل في RPE65. تشكل طفرات RPE65 حوالي 8% من جميع حالات LCA، وبالتالي فإن الغالبية العظمى من المرضى غير مؤهلين. العلاجات للأنماط الجينية الأخرى لا تزال في مرحلة البحث.
Qكيف يمكن الحصول على العلاج الجيني في اليابان؟
A
يشترط تأكيد وجود طفرات ثنائية الأليل في RPE65 ووجود عدد كافٍ من خلايا الشبكية الحية. الخطوة الأولى هي التشخيص الجيني باستخدام لوحة الجينات (PrismGuide IRD panel). يُوصى بالتحويل إلى مراكز متخصصة لديها خبرة في علاج أمراض الشبكية الوراثية.
ترتبط الفيزيولوجيا المرضية لـ LCA بتعطل الدورة البصرية (Visual Cycle)، مما يمنع العين من نقل المعلومات الضوئية.
الدورة البصرية هي سلسلة من التفاعلات الأنزيمية بين الظهارة الصبغية للشبكية (RPE) والشبكية العصبية الحسية، حيث يتم استقلاب فيتامين A الغذائي لإنتاج 11-سي-ريتينال (11-cis retinal) اللازم لتكوين الصبغات البصرية. بدون 11-سي-ريتينال، لا تبدأ سلسلة نقل الإشارات الضوئية، ولا تنتقل الإشارات العصبية البصرية إلى القشرة البصرية. تؤدي الطفرات في أي من الجينات المشفرة للبروتينات المشاركة في هذه السلسلة إلى تعطل الدورة البصرية وظهور أعراض LCA.
من الناحية النسيجية المرضية، هناك دليل على تورط الشبكية الخارجية والمستقبلات الضوئية، مما يشير إلى أن LCA هو عملية تنكسية وليس خلل تكويني.
GUCY2D (LCA1): يرمز لإنزيم غوانيلات سيكلاز الخاص بالشبكية (GC-E). يحفز تخليق cGMP، وهو مفتاح نقل الإشارات الضوئية. تم تحديد أكثر من 140 طفرة مرتبطة بالمرض، 88% منها تسبب LCA متنحي جسدي. الحمض الأميني 838 معروف كنقطة ساخنة للطفرات 2).
RPE65 (LCA2): ينتمي إلى عائلة أوكسجيناز قطع الكاروتينويد، وهو إنزيم ثنائي الوظيفة يحفز قطع إستر O-ألكيل من all-trans-retinyl ester وأيزومرة جزء الريتينيل (all-trans-retinol → 11-cis-retinol) 2). ضروري لوظائف كل من العصي والمخاريط، وتشير الدراسات الحديثة إلى احتمالية مشاركته في أيزومرة اللوتين إلى ميسو-زياكسانثين 2). العلاج الجيني الوحيد المعتمد له.
CRB1 (LCA8): متماثل مع بروتين crumbs في ذبابة الفاكهة، ويُعبر عنه في الجزء الداخلي من المستقبلات الضوئية وخلايا مولر. مهم للحفاظ على الاستقطاب الخلوي، ويقع على الكروموسوم 1q31.3 1).
CEP290 (LCA10): يشارك في وظيفة الأهداب للمستقبلات الضوئية. أعلى تواتر للطفرات بين جينات LCA (حوالي 15%).
NMNAT1 (LCA9): يرمز للإنزيم الرئيسي في تخليق NAD (نيكوتيناميد أدينين ثنائي النوكليوتيد) 2).
LCA5: يرمز لبروتين ليبرسيلي (lebercilin)، ويشارك في وظيفة الأهداب والنقل البروتيني داخل الأهداب 2).
AIPL1: يعمل كمرافق خاص لـ PDE6 (فوسفودايستراز 6). يؤدي نقص AIPL1 إلى عدم استقرار PDE6 → خلل في استقلاب cGMP → شذوذ في القنوات → تنكس المستقبلات الضوئية 2).
7. أحدث الأبحاث والتوجهات المستقبلية (تقارير المرحلة البحثية)
في التقييم طويل المدى لـ فوريتيجين نيبيرفوفيك (NCT00481546، NCT00643747)، تم الإبلاغ عن ذروة أولية بعد 6-12 شهرًا من العلاج، ثم انخفاض تدريجي في الفوائد السريرية بما في ذلك حساسية الشبكية وحدة البصر والمكاسب الوظيفية 6). في دراسة PERCEIVE (دراسة تسجيلية استباقية)، تم الإبلاغ عن بيانات السلامة والفعالية لمدة عامين في الممارسة السريرية الحقيقية، ولوحظ التهاب العنبية المرتبط بالعلاج الجيني (GTAU) في ما يصل إلى 50% من الحالات 9).
طورته شركة Editas Medicine. يحمل ناقل AAV5 Cas9 من المكورات العنقودية الذهبية واثنين من RNAs الدليل، ويستهدف طفرة داخل الإنترون العميق (c.2991+1655A>G) الموجودة في الإنترون 26 من جين CEP290 3). تم تأكيد السلامة في أول تجربة على البشر، وأظهرت تحملاً جيدًا حتى عند الجرعات العالية نسبيًا 3).
استعادة الوظيفة البصرية عن طريق علم البصريات الوراثي (Optogenetics)
كمنهج مستقل عن الجينات، يتم دراسة تقنية التعبير عن قنوات الرودوبسين الحساسة للضوء في الخلايا العصبية الشبكية الداخلية المتبقية 3). قد تكون قابلة للتطبيق على جميع أنواع LCA (بغض النظر عن النمط الجيني)، وتجارب سريرية مبكرة جارية.
Duan W, Zhou T, Jiang H, Zhang M, Hu M, Zhang L. A novel nonsense variant (c.1499C>G) in CRB1 caused Leber congenital amaurosis-8 in a Chinese family and a literature review. BMC Med Genomics. 2022;15(1):197. doi:10.1186/s12920-022-01356-z.
Mordà D, Alibrandi S, Scimone C, Rinaldi C, Scalinci SZ, Abate G, et al. Decoding pediatric inherited retinal dystrophies: Bridging genetic complexity and clinical heterogeneity. Progress in retinal and eye research. 2025;109:101405. doi:10.1016/j.preteyeres.2025.101405. PMID:40998214.
Botto C, Rucli M, Tekinsoy MD, Pulman J, Sahel JA, Dalkara D. Early and late stage gene therapy interventions for inherited retinal degenerations. Progress in retinal and eye research. 2022;86:100975. doi:10.1016/j.preteyeres.2021.100975. PMID:34058340.
Gurnani B, et al. Clinical approach to pediatric nystagmus: a comprehensive diagnostic algorithm. Clin Ophthalmol. 2025;19:1617-1636.
Russell S, Bennett J, Wellman JA, Chung DC, Yu ZF, Tillman A, et al. Efficacy and safety of voretigene neparvovec (AAV2-hRPE65v2) in patients with RPE65-mediated inherited retinal dystrophy: a randomised, controlled, open-label, phase 3 trial. Lancet (London, England). 2017;390(10097):849-860. doi:10.1016/S0140-6736(17)31868-8. PMID:28712537; PMCID:PMC5726391.
Maguire AM, Russell S, Wellman JA, Chung DC, Yu ZF, Tillman A, et al. Efficacy, Safety, and Durability of Voretigene Neparvovec-rzyl in RPE65 Mutation-Associated Inherited Retinal Dystrophy: Results of Phase 1 and 3 Trials. Ophthalmology. 2019;126(9):1273-1285. doi:10.1016/j.ophtha.2019.06.017. PMID:31443789.
Fujinami K, Akiyama K, Tsunoda K, et al. Efficacy and Safety of Voretigene Neparvovec in RPE65-Retinopathy: Results of a Phase III Trial in Japan. Ophthalmol Sci. 2025;5:100876. doi:10.1016/j.xops.2025.100876. PMID:40910102; PMCID:PMC12405627.
Gange WS, Sisk RA, Besirli CG, Lee TC, Havunjian M, Schwartz H, et al. Perifoveal Chorioretinal Atrophy after Subretinal Voretigene Neparvovec-rzyl for RPE65-Mediated Leber Congenital Amaurosis. Ophthalmology. Retina. 2022;6(1):58-64. doi:10.1016/j.oret.2021.03.016. PMID:33838313; PMCID:PMC8497635.
Fischer MD, Simonelli F, Sahni J, Holz FG, Maier R, Fasser C, et al. Real-World Safety and Effectiveness of Voretigene Neparvovec: Results up to 2 Years from the Prospective, Registry-Based PERCEIVE Study. Biomolecules. 2024;14(1). doi:10.3390/biom14010122. PMID:38254722; PMCID:PMC10813228.
انسخ نص المقال والصقه في مساعد الذكاء الاصطناعي الذي تفضله.
تم نسخ المقال إلى الحافظة
افتح أحد مساعدي الذكاء الاصطناعي أدناه والصق النص المنسوخ في مربع المحادثة.