레버 선천성 흑암시(Leber congenital amaurosis; LCA)는 출생 시~유아기에 중증 시각 장애를 초래하는 선천성 망막 이영양증 중 가장 중증인 형태입니다. 소아 시각 장애의 주요 원인 질환이며, 선천성 시각 장애의 대표적 원인 질환으로 알려져 있습니다. 임상 양상은 다양합니다.
1869년 독일의 안과 의사 테오도르 카를 구스타프 폰 레버(Theodor Karl Gustav von Leber, 1840–1917)에 의해 처음 보고되었습니다. 같은 레버가 1871년에 보고한 레버 유전성 시신경병증(LHON)은 20세 전후에 발병하는 미토콘드리아 질환으로, LCA와는 전혀 다른 질환입니다. 1957년 망막전위도(ERG)에서 파형 소실이 LCA 진단의 공통 특징으로 확인되어 질환명이 확립되었습니다.
추정 출생 유병률은 출생 10만 명당 23명(1/30,0001/81,000)입니다1). 보고에 따라 1:80,0001:200,000으로 하는 문헌도 있어 폭이 있습니다2). 전체 망막 이영양증의 약 5%를 차지하며, 맹학교에 다니는 시각 장애 아동의 약 20%가 LCA입니다1). 현재 약 27개의 LCA 관련 유전자가 동정되었으며2), 증례의 약 7080%에서 원인 유전자가 특정됩니다2). 유전 양식은 상염색체 열성 유전이 주이나, 우성 유전이나 X연관 유전의 보고도 있습니다.
Q레버 선천성 흑암시는 언제쯤 알 수 있나요?
A
보통 생후 6주경에 안진이나 주시 결여로 부모가 알아차리는 경우가 많습니다3). 중증의 시각 반응 불량(주시·추시가 전혀 안 됨)이 인정된 경우 LCA를 의심하고 망막전위도로 확진합니다.
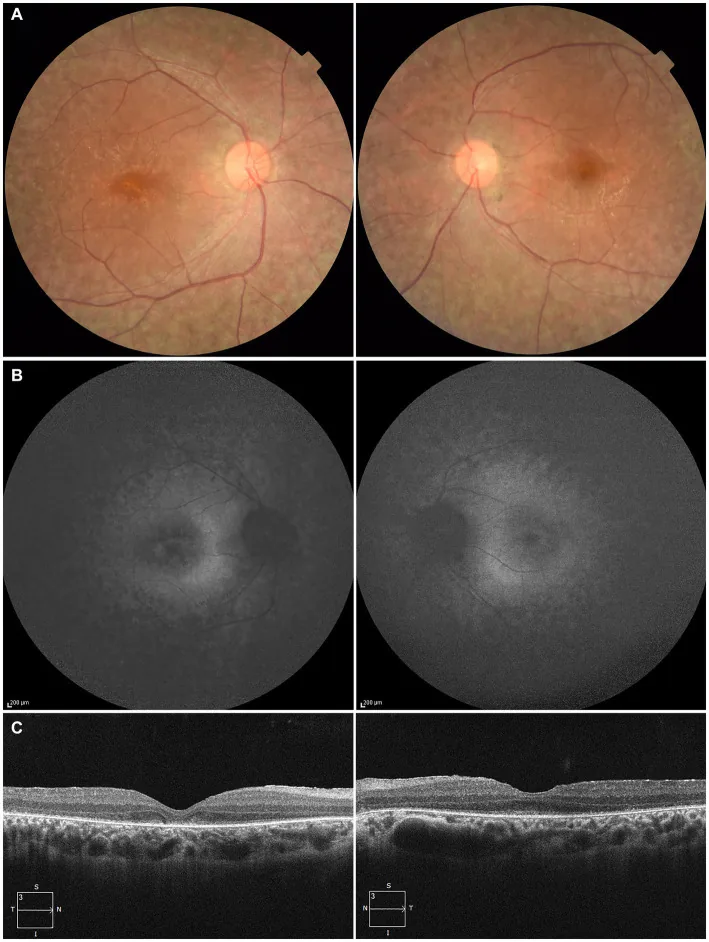
Higa N, et al. A novel RPE65 variant p.(Ala391Asp) in Leber congenital amaurosis: a case report and literature review in Japan. Front Med (Lausanne). 2024. Figure 1. PMCID: PMC11446184. License: CC BY.
27세 환자의 양안: (A) 안저 사진에서 망막색소상피 탈색소와 망막혈관 협소화; (B) 안저 자가형광에서 병변 부위의 과형광과 신호 감소; (C) OCT에서 우안의 타원체대(EZ) 소실과 좌안 중심와의 미약한 잔존. 본문 “2. 주요 증상과 임상 소견”에서 다루는 망막색소상피 변성과 망막혈관 협소화에 해당함.
영아기에는 안저가 정상으로 보이는 경우가 많지만, 이후 다양한 소견이 나타납니다. 안저 소견은 정상 안저에서 전형적인 망막색소변성증 양상까지 다양합니다. 진행된 증례에서는 시신경 유두가 창백해지고 혈관이 매우 협소해지며, 안저 전체의 색조가 어두워지고 황반의 윤상 반사가 소실됩니다.
일본 코호트(34가계)의 NGS 분석에서 검출률은 약 56%였으며, 가장 빈번한 돌연변이 유전자는 CRB1, NMNAT1, RPGRIP1로 보고되었습니다1).
원인 유전자의 동정은 치료 적격성 판단에 직접 연결됩니다. 특히 RPE65 돌연변이 확인은 유전자 치료(voretigene neparvovec)의 적응 판정에 필수적입니다2).
AIPL1은 포스포디에스테라제 6(PDE6)의 특수 샤페론으로 기능하며, PDE6는 광정보 전달에서 cGMP 분해를 담당합니다. AIPL1 결핍은 PDE6 단백질 양을 급격히 감소시켜 cGMP 대사 붕괴 → 광수용체 변성 → 조기 실명으로 이어집니다2). AIPL1 돌연변이는 LCA 전체의 약 5~10%를 차지합니다2).
Q다음 아이도 LCA가 유전될 확률은?
A
상염색체 열성 유전의 경우, 부모가 모두 보인자라면 다음 아이가 발병할 확률은 25%, 보인자가 될 확률은 50%, 발병하지 않고 보인자가 아닐 확률은 25%입니다. 원인 유전자가 확인되면 산전 진단이나 착상 전 유전 진단도 선택지가 됩니다.
유전자 검사: 차세대 염기서열 분석(NGS), DNA 마이크로어레이, 연쇄 분석 등이 사용됩니다. 전체 검출률은 약 70~80%입니다2). 2023년부터 82개의 IRD 원인 유전자에 대한 패널 검사(PrismGuide IRD 패널)가 건강보험에 포함되어, RPE65 관련 IRD가 의심되는 젊은 발병 환자에게 적용됩니다.
2017년, 미국 FDA는 양쪽 대립유전자 RPE65 변이와 관련된 LCA2의 치료제로 **보레티진 네파르보벡(voretigene neparvovec-rzyl; 상품명 Luxturna)**을 승인했습니다. 이는 안과 분야에서 최초로 FDA 승인을 받은 유전자 치료 제품입니다. 2023년에는 일본에서도 승인되었습니다(제품명: Luxturna® 주사제).
재조합 아데노연관바이러스(rAAV2) 벡터를 통해 정상 RPE65 유전자 사본을 망막색소상피에 망막하 주사로 도입합니다. 유리체 수술실에서 시행됩니다.
3상 301 시험 (Russell 2017)5):
대상: 31명의 RPE65 관련 IRD 환자
1차 평가변수: MLMT(다중 조도 이동성 검사; 다양한 조도에서의 행동 관찰 검사)
전시야 자극 역치(FST)에서도 유의한 개선이 관찰됨
1/3상 장기 결과 (Maguire 2019)6):
치료 후 6~12개월에 최고치에 도달한 망막 민감도, 시력, 기능적 이득이 이후 점진적으로 감소하는 경향이 나타남
그러나 RPE65 돌연변이는 전체 LCA 환자의 약 8%에 불과합니다. 다른 돌연변이 유형에 대해서는 현재 효과가 입증된 치료법이 없습니다.
Q유전자 치료는 모든 LCA 환자에게 사용할 수 있나요?
A
현재 승인된 유전자 치료(보레티진 네파르보벡 / 룩스터나® 주사)의 적응증은 양쪽 대립유전자 RPE65 돌연변이로 인한 LCA2에 국한됩니다. RPE65 돌연변이는 전체 LCA의 약 8%를 차지하므로 대부분의 환자에게는 적응증이 없습니다. 다른 유전자형에 대한 치료는 연구 단계에 있습니다.
Q일본에서 유전자 치료를 받으려면 어떻게 해야 하나요?
A
양쪽 대립유전자 RPE65 돌연변이가 확인되고 충분한 생존 망막 세포가 있어야 합니다. 유전자 패널 검사(PrismGuide IRD 패널)를 통한 유전자 진단이 첫 단계입니다. 유전성 망막 질환 진료 경험이 있는 전문 기관으로의 의뢰가 권장됩니다.
LCA의 병태생리는 시각 순환(Visual Cycle)의 파괴로 인해 눈이 빛 정보를 전달할 수 없는 것과 관련이 있습니다.
시각 순환은 망막색소상피(RPE)와 신경감각망막 사이에서 일어나는 일련의 효소 반응으로, 식이 비타민 A를 대사하여 11-시스-레티날(11-cis retinal)을 생성하고 광색소를 생성합니다. 11-시스-레티날이 없으면 광전달 연쇄반응이 시작되지 않아 시각 신경 신호가 시각 피질로 전달되지 않습니다. 이 연쇄반응에 관여하는 단백질을 코딩하는 유전자 중 하나에 돌연변이가 있으면 시각 순환이 차단되어 LCA 증상이 나타납니다.
조직병리학적으로 외망막과 광수용체의 관여가 입증되어 LCA가 형성 부전이 아닌 퇴행성 과정임을 시사합니다.
GUCY2D (LCA1): 망막 특이적 구아닐산 시클라제(GC-E)를 코딩합니다. cGMP 합성을 촉매하며 광신호 전달의 핵심입니다. 140개 이상의 질병 관련 돌연변이가 확인되었으며, 88%가 상염색체 열성 LCA의 원인입니다. 838번 아미노산은 돌연변이 핫스팟으로 알려져 있습니다2).
RPE65 (LCA2): 카로티노이드 절단 산소화효소 슈퍼패밀리에 속합니다. all-trans-레티닐 에스테르로부터 O-알킬 에스테르 절단과 레티닐 부분의 이성질화(all-trans-레티놀 → 11-cis-레티놀)를 촉매하는 이중 기능 효소입니다2). 간상체와 원추체 기능 모두에 필수적이며, 최근 연구에서는 루테인에서 메조-제아잔틴으로의 이성질화에도 관여할 가능성이 제시되었습니다2). 유전자 치료의 유일한 승인 적응증입니다.
보레티진 네파르보벡의 장기 추적 평가(NCT00481546, NCT00643747)에서는 치료 후 6~12개월에 초기 피크가 나타난 후, 망막 감도, 시력, 기능적 이득을 포함한 임상적 이점이 점진적으로 감소하는 것으로 보고되었습니다6). PERCEIVE 연구(전향적 등록 연구)에서는 실제 임상에서의 2년간 안전성 및 유효성 데이터가 보고되었으며, 최대 50%의 증례에서 GTAU(유전자 치료 관련 포도막염)가 인정되었습니다9).
LCA의 경과는 안정(약 75%), 진행성 악화(약 15%), 개선(약 10%)의 세 가지 패턴으로 분류된다. AIPL1 돌연변이는 진행성 악화와, RPGRIP1 돌연변이는 안정적인 경과와 관련된다. 미래에는 인공 시각, 유전자 치료, 재생 의학 등을 통한 진행 중단이나 치료가 기대된다.
Duan W, Zhou T, Jiang H, Zhang M, Hu M, Zhang L. A novel nonsense variant (c.1499C>G) in CRB1 caused Leber congenital amaurosis-8 in a Chinese family and a literature review. BMC Med Genomics. 2022;15(1):197. doi:10.1186/s12920-022-01356-z.
Mordà D, Alibrandi S, Scimone C, Rinaldi C, Scalinci SZ, Abate G, et al. Decoding pediatric inherited retinal dystrophies: Bridging genetic complexity and clinical heterogeneity. Progress in retinal and eye research. 2025;109:101405. doi:10.1016/j.preteyeres.2025.101405. PMID:40998214.
Botto C, Rucli M, Tekinsoy MD, Pulman J, Sahel JA, Dalkara D. Early and late stage gene therapy interventions for inherited retinal degenerations. Progress in retinal and eye research. 2022;86:100975. doi:10.1016/j.preteyeres.2021.100975. PMID:34058340.
Gurnani B, et al. Clinical approach to pediatric nystagmus: a comprehensive diagnostic algorithm. Clin Ophthalmol. 2025;19:1617-1636.
Russell S, Bennett J, Wellman JA, Chung DC, Yu ZF, Tillman A, et al. Efficacy and safety of voretigene neparvovec (AAV2-hRPE65v2) in patients with RPE65-mediated inherited retinal dystrophy: a randomised, controlled, open-label, phase 3 trial. Lancet (London, England). 2017;390(10097):849-860. doi:10.1016/S0140-6736(17)31868-8. PMID:28712537; PMCID:PMC5726391.
Maguire AM, Russell S, Wellman JA, Chung DC, Yu ZF, Tillman A, et al. Efficacy, Safety, and Durability of Voretigene Neparvovec-rzyl in RPE65 Mutation-Associated Inherited Retinal Dystrophy: Results of Phase 1 and 3 Trials. Ophthalmology. 2019;126(9):1273-1285. doi:10.1016/j.ophtha.2019.06.017. PMID:31443789.
Fujinami K, Akiyama K, Tsunoda K, et al. Efficacy and Safety of Voretigene Neparvovec in RPE65-Retinopathy: Results of a Phase III Trial in Japan. Ophthalmol Sci. 2025;5:100876. doi:10.1016/j.xops.2025.100876. PMID:40910102; PMCID:PMC12405627.
Gange WS, Sisk RA, Besirli CG, Lee TC, Havunjian M, Schwartz H, et al. Perifoveal Chorioretinal Atrophy after Subretinal Voretigene Neparvovec-rzyl for RPE65-Mediated Leber Congenital Amaurosis. Ophthalmology. Retina. 2022;6(1):58-64. doi:10.1016/j.oret.2021.03.016. PMID:33838313; PMCID:PMC8497635.
Fischer MD, Simonelli F, Sahni J, Holz FG, Maier R, Fasser C, et al. Real-World Safety and Effectiveness of Voretigene Neparvovec: Results up to 2 Years from the Prospective, Registry-Based PERCEIVE Study. Biomolecules. 2024;14(1). doi:10.3390/biom14010122. PMID:38254722; PMCID:PMC10813228.