โรคจอประสาทตา เสื่อมแต่กำเนิดชนิดเลเบอร์ (LCA) เป็นรูปแบบที่รุนแรงที่สุดของโรคจอประสาทตา เสื่อมแต่กำเนิด ทำให้เกิดความบกพร่องทางการมองเห็น อย่างรุนแรงตั้งแต่แรกเกิดถึงวัยทารก
ส่วนใหญ่ถ่ายทอดทางพันธุกรรมแบบ autosomal recessive และมีการระบุยีนก่อโรคประมาณ 27 ยีนแล้ว
อาการตากระตุก (nystagmus) ปฏิกิริยารูม่านตา ลดลงหรือหายไป และสัญญาณนิ้ว-ตา (oculodigital sign) เป็นลักษณะทางคลินิกที่เด่นชัด
การตรวจคลื่นไฟฟ้าจอประสาทตา (ERG ) ที่พบว่าการตอบสนองของเซลล์รูปแท่ง และเซลล์รูปกรวย หายไปหรือลดลงอย่างมากเป็นกุญแจสำคัญในการวินิจฉัย
ความคมชัดของการมองเห็น ของผู้ป่วยส่วนใหญ่อยู่ต่ำกว่า 0.1 และประมาณหนึ่งในสามไม่มีการรับรู้แสง
การรักษาด้วยยีน (voretigene neparvovec/Luxturna®) ได้รับการอนุมัติในญี่ปุ่นในปี 2023 สำหรับ LCA2 ที่เกิดจากการกลายพันธุ์ของยีน RPE 65 แต่ครอบคลุมเพียงประมาณ 8% ของผู้ป่วย LCA ทั้งหมด
สำหรับชนิดอื่นๆ ยังไม่มีการรักษาที่มีประสิทธิภาพที่ได้รับการยอมรับ การดูแลจะเน้นที่การแก้ไขค่าสายตา การฝึกการมองเห็น และการดูแลผู้มีความบกพร่องทางการเห็น
โรคจอประสาทตา เสื่อมแต่กำเนิดชนิดเลเบอร์ (Leber congenital amaurosis; LCA) เป็นรูปแบบที่รุนแรงที่สุดของโรคจอประสาทตา เสื่อมแต่กำเนิด ทำให้เกิดความบกพร่องทางการมองเห็น อย่างรุนแรงตั้งแต่แรกเกิดถึงวัยทารก เป็นสาเหตุหลักของความบกพร่องทางการมองเห็น ในเด็ก และเป็นที่รู้จักในฐานะสาเหตุทั่วไปของตาบอดแต่กำเนิด ลักษณะทางคลินิกมีความหลากหลาย
รายงานครั้งแรกในปี ค.ศ. 1869 โดยจักษุแพทย์ชาวเยอรมัน Theodor Karl Gustav von Leber (1840–1917) หมายเหตุ: โรคเส้นประสาทตา ฝ่อชนิดเลเบอร์ (LHON ) ซึ่งรายงานโดยเลเบอร์คนเดียวกันในปี ค.ศ. 1871 เป็นโรคไมโทคอนเดรียที่เริ่มมีอาการในช่วงอายุประมาณ 20 ปี และแตกต่างจาก LCA โดยสิ้นเชิง ในปี ค.ศ. 1957 การหายไปของคลื่นในการตรวจ ERG ได้รับการยืนยันว่าเป็นลักษณะร่วมในการวินิจฉัย LCA ซึ่งนำไปสู่การกำหนดชื่อโรค
ความชุกโดยประมาณเมื่อแรกเกิดคือ 2–3 คนต่อการเกิด 100,000 คน (1/30,000 ถึง 1/81,000)1) รายงานบางฉบับระบุ 1:80,000 ถึง 1:200,000 ซึ่งแสดงถึงความแปรผัน2) LCA คิดเป็นประมาณ 5% ของโรคจอประสาทตา เสื่อมทั้งหมด และประมาณ 20% ของเด็กที่มีความบกพร่องทางการเห็นในโรงเรียนคนตาบอดเป็นผู้ป่วย LCA1) ปัจจุบันมีการระบุยีนที่เกี่ยวข้องกับ LCA ประมาณ 27 ยีน2) และพบยีนก่อโรคในประมาณ 70–80% ของกรณี2) รูปแบบการถ่ายทอดทางพันธุกรรมหลักคือ autosomal recessive แต่มีรายงานการถ่ายทอดแบบ autosomal dominant และ X-linked
Q
โรคจอประสาทตาเสื่อมแต่กำเนิดชนิดเลเบอร์สามารถตรวจพบได้เมื่อใด?
A
โดยปกติผู้ปกครองจะสังเกตเห็นอาการตากระตุก หรือการไม่จ้องมองเมื่ออายุประมาณ 6 สัปดาห์3) หากมีการตอบสนองทางการมองเห็น ที่ไม่ดี (ไม่สามารถจ้องหรือติดตามวัตถุได้) จะสงสัย LCA และยืนยันการวินิจฉัยด้วย ERG
Higa N, et al. A novel
RPE 65 variant p.(Ala391Asp) in Leber congenital amaurosis: a case report and literature review in Japan. Front Med (Lausanne). 2024. Figure 1. PM
CI D: PMC11446184. License: CC BY.
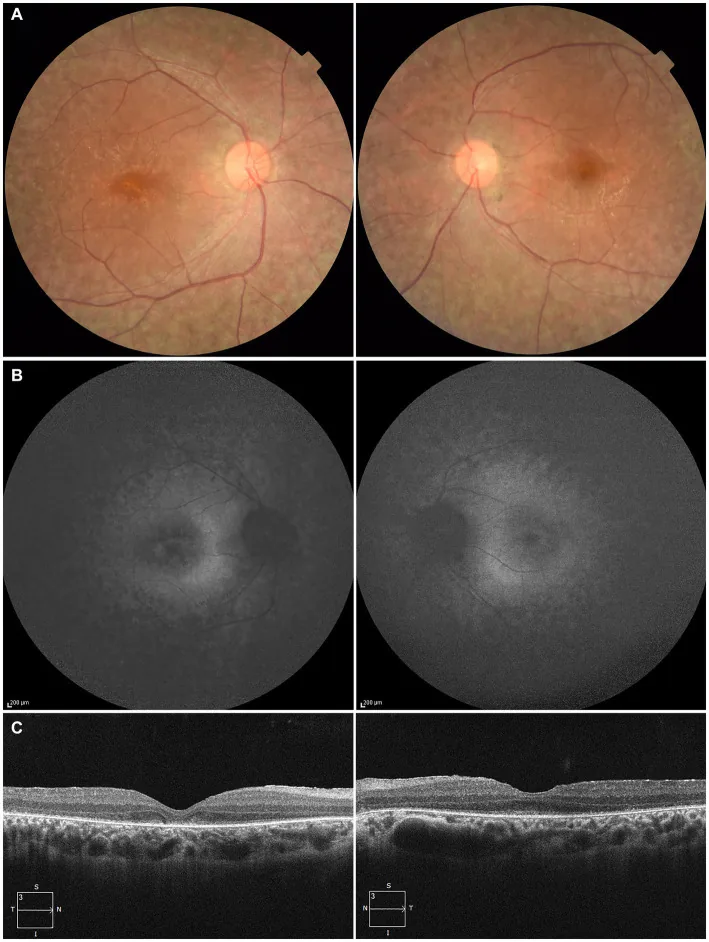
ในผู้ป่วยอายุ 27 ปี (A) ภาพถ่ายจอประสาทตา ทั้งสองข้างแสดงการสูญเสียเม็ดสีของเยื่อบุผิวรงควัตถุจอประสาทตา และการตีบของหลอดเลือดจอประสาทตา (B) การเรืองแสงเองของจอประสาทตา แสดงการเรืองแสงมากเกินไปและสัญญาณลดลงในรอยโรค (C) OCT แสดงการหายไปของโซนอิลลิปซอยด์ (EZ ) ในตาขวาและเหลือเพียงเล็กน้อยในรอยบุ๋มจอประสาทตา ซ้าย สอดคล้องกับความเสื่อมของเยื่อบุผิวรงควัตถุจอประสาทตา และการตีบของหลอดเลือดจอประสาทตา ที่กล่าวถึงในหัวข้อ “2. อาการหลักและอาการแสดงทางคลินิก”
มีความบกพร่องทางการมองเห็น อย่างรุนแรงตั้งแต่แรกเกิดหรือช่วงหลังคลอดตอนต้น
การมองเห็น ลดลงการมองเห็น ต่ำกว่า 0.1 และประมาณหนึ่งในสามไม่มีการรับรู้แสง ความบกพร่องทางการมองเห็น โดยทั่วไปคงที่หรือดำเนินไปช้ามากกลัวแสง : หลายกรณีแสดงอาการไวต่อแสง มากเกินไปตาบอดกลางคืน การมองเห็น ในที่มืดแย่ลงไปอีก
บ่อยครั้งที่ผู้ปกครองสังเกตเห็นอาการตากระตุก หรือการไม่จ้องมองเมื่ออายุประมาณ 6 สัปดาห์3)
ตากระตุก จอประสาทตา เสื่อมชนิดรุนแรงที่เริ่มต้นเร็ว (EOSRD) ซึ่ง EOSRD จะไม่มีตากระตุก 2) ความผิดปกติของปฏิกิริยารูม่านตา : รีเฟล็กซ์ต่อแสงช้าหรือหายไป เรียกว่า “รูม่านตา บอด (amaurotic pupils)”สัญญาณตา-นิ้ว (oculo-digital sign) : การกระทำที่ใช้นิ้วแหย่ กด หรือขยี้ตา ซึ่งเชื่อว่าเป็นความพยายามกระตุ้นจอประสาทตา โดยกลไกเพื่อให้เกิดการมองเห็น ผลกระทบหลักคือลูกตาลึก (enophthalmos) เนื่องจากการฝ่อของไขมันในเบ้าตา ความผิดปกติของการหักเหแสง : สายตายาว มาก (>5 ไดออปเตอร์) พบได้บ่อย เชื่อว่าเกิดจากความล้มเหลวของกระบวนการทำให้สายตาปกติ (emmetropization) เนื่องจากความบกพร่องทางการมองเห็น ตั้งแต่แรกเริ่ม
ในวัยทารก จอประสาทตา มักดูปกติ แต่ต่อมาจะมีอาการแสดงที่หลากหลาย อาการแสดงของจอประสาทตา มีตั้งแต่จอประสาทตา ปกติไปจนถึงจอประสาทตา ที่มีลักษณะคล้ายจอประสาทตา อักเสบชนิดรงควัตถุ (retinitis pigmentosa) ทั่วไป ในกรณีที่รุนแรง หัวประสาทตาจะซีด หลอดเลือดตีบแคบมาก สีโดยรวมของจอประสาทตา มืดลง และรีเฟล็กซ์วงแหวนรอบรอยบุ๋มจอประสาทตา หายไป
จานประสาทตา ซีดหลอดเลือดจอตาตีบแคบ จอตาแบบเกลือพริกไทย : จอประสาทตา เสื่อมของชั้น pigment epithelium มีจุดขาวเล็กๆ และการสูญเสียสีเม็ดสีรูปกระดูกสันหลัง และ จุดใต้จอตา (จอตาลายหินอ่อน) จุดภาพชัด เสื่อมสารคัดหลั่งคล้ายโรค Coats รอยโรคคล้ายเหรียญมีสี
ในการตรวจ OCT ชั้นนอกของจอตาเกือบหายไป โดยมีลักษณะบางลงจนหายไปของ ellipsoid zone (IS/OS junction) ผลการตรวจแตกต่างกันตาม genotype:
การกลายพันธุ์ CRB1 : ความหนาจอตาเพิ่มขึ้นอย่างขัดแย้ง (coarse lamination) 1)
การกลายพันธุ์ RPE 65FAF (การเรืองแสงเองของจอตา) หายไป 2)
การกลายพันธุ์ GUCY2D : FAF ปกติ 2)
การกลายพันธุ์ NMNAT1 : จุดภาพชัด ฝ่อ (macular coloboma-like atrophy)
ภาวะแทรกซ้อน เช่น กระจกตา รูปกรวย ต้อกระจก และต้อหิน เพิ่มขึ้นตามอายุ
LCA แบ่งเป็น ชนิดธรรมดา ที่มีเฉพาะอาการทางตา และ ชนิดซับซ้อน ที่มีโรคร่วมทั่วร่างกาย
ชนิดธรรมดา
อาการทางตาเท่านั้น : ความบกพร่องทางการมองเห็น เป็นหลัก ไม่มีความผิดปกติทั่วร่างกาย
สายตายาว ปานกลางถึงรุนแรง
ชนิดซับซ้อน
ความผิดปกติของระบบประสาทส่วนกลาง : สมองน้อย vermis เจริญไม่เต็มที่, ความผิดปกติของก้านสมอง
พัฒนาการทางจิตล่าช้า : บางกรณีมีความบกพร่องทางสติปัญญาร่วมด้วย
โรคไต : อาจมีโรคถุงน้ำในไตร่วมด้วย
อื่นๆ : สูญเสียการได้ยิน, ความผิดปกติของโครงกระดูก, โรคตับ, ความผิดปกติของเมตาบอลิซึม, โรคลมชัก
Q
สัญญาณการขยี้ตา (การเคลื่อนไหวขยี้ตา) มีจุดประสงค์เพื่ออะไร?
A
พฤติกรรมเฉพาะของผู้ป่วย LCA คือการใช้นิ้วจิ้มหรือกดตาเพื่อกระตุ้นจอประสาทตา โดยกลไกและพยายามทำให้เกิดการมองเห็น การทำซ้ำเป็นเวลานานทำให้ไขมันในเบ้าตา ฝ่อ ส่งผลให้ตาลึก
LCA เป็นกลุ่มโรคจอประสาทตา เสื่อมที่ถ่ายทอดทางพันธุกรรม ส่วนใหญ่เป็นแบบถ่ายทอดทางยีนด้อยบนออโตโซม2) พบน้อยที่เกิดจากยีนเด่นบนออโตโซมจากการกลายพันธุ์ของ CRX , IMPDH1 และ OTX2 2) นอกจากนี้ยังมีรายงานการถ่ายทอดแบบเชื่อมโยงกับโครโมโซม X
ปัจจุบันมีการรายงาน LCA 19 ชนิดตั้งแต่ LCA1 ถึง LCA19 รวมถึงยีนที่เกี่ยวข้องอีก 8 ยีน2) ยีนก่อโรคเกี่ยวข้องกับหลายวิถีทางที่เกี่ยวกับการพัฒนาและการทำงานของจอประสาทตา เช่น การสร้างรูปร่างของเซลล์รับแสง , ซีเลียที่นำสัญญาณแสง, และวงจรการมองเห็น
ยีนที่เกี่ยวข้องกับ LCA ถูกจำแนกออกเป็น 5 เครือข่ายการทำงานหลัก2) :
เมแทบอลิซึมของเรตินอยด์และวงจรการมองเห็น ของเซลล์รูปแท่ง (RPE 65, LRAT, RDH12)การรักษาสมดุลของจอประสาทตา และการรักษาเซลล์รับแสง (AIPL1, SPATA7, TULP1, USP45, CRB1, LCA5)การพัฒนาจอประสาทตา และการสร้างรูปร่าง (RD3, CEP290)การตรวจจับสิ่งกระตุ้นแสงและการรับรู้ทางสายตา (GUCY2D, CNGA3)ซิเลียเชื่อมต่อเซลล์รับแสง และการรักษาส่วนนอก (CEP290, RP GRIP1, RP GR)
ยีนที่กลายพันธุ์บ่อยที่สุดในโลกและสัดส่วนมีดังนี้:
ยีน สัดส่วน วิถีที่เกี่ยวข้อง CEP290 ประมาณ 15% การทำงานของซิเลีย GUCY2D ประมาณ 12% การส่งสัญญาณแสง (การสังเคราะห์ cGMP) CRB1 ประมาณ 10% การรักษาขั้วของเซลล์ RPE 65ประมาณ 8% เมแทบอลิซึมของเรตินอยด์
ในกลุ่มตัวอย่างชาวญี่ปุ่น (34 ครอบครัว) การวิเคราะห์ NGS แสดงอัตราการตรวจพบประมาณ 56% และยีนที่กลายพันธุ์บ่อยที่สุดที่รายงานคือ CRB1 , NMNAT1 และ RP GRIP11) .
การระบุยีนที่เป็นสาเหตุเกี่ยวข้องโดยตรงกับการพิจารณาความเหมาะสมในการรักษา โดยเฉพาะอย่างยิ่ง การยืนยันการกลายพันธุ์ของ RPE 652) .
AIPL1 ทำหน้าที่เป็น chaperone เฉพาะสำหรับฟอสโฟไดเอสเทอเรส 6 (PDE6) ซึ่งย่อยสลาย cGMP ในการถ่ายทอดสัญญาณแสง การขาด AIPL1 ทำให้ปริมาณโปรตีน PDE6 ลดลงอย่างรุนแรง นำไปสู่ความล้มเหลวของเมแทบอลิซึมของ cGMP → การเสื่อมของเซลล์รับแสง → ตาบอดเร็ว2) การกลายพันธุ์ของ AIPL1 คิดเป็นประมาณ 5-10% ของผู้ป่วย LCA ทั้งหมด2) .
การให้คำปรึกษาทางพันธุกรรม
ในกรณีของการถ่ายทอดทางพันธุกรรมแบบ autosomal recessive พี่น้องของเด็กที่ป่วยมีโอกาส 25% ที่จะป่วย และ 50% ที่จะเป็นพาหะไม่มีอาการ หากทราบการกลายพันธุ์ที่เป็นสาเหตุ สามารถตรวจหาพาหะและการวินิจฉัยก่อนคลอดได้ แนะนำให้รับคำปรึกษาทางพันธุกรรม
Q
ความน่าจะเป็นที่บุตรคนต่อไปจะเป็น LCA คือเท่าใด?
A
ในการถ่ายทอดทางพันธุกรรมแบบ autosomal recessive หากทั้งพ่อและแม่เป็นพาหะ ความน่าจะเป็นที่บุตรคนต่อไปจะป่วยคือ 25% ความน่าจะเป็นที่จะเป็นพาหะคือ 50% และความน่าจะเป็นที่จะไม่ป่วยและไม่เป็นพาหะคือ 25% หากทราบยีนที่เป็นสาเหตุ การวินิจฉัยก่อนคลอดหรือการวินิจฉัยก่อนการฝังตัวของตัวอ่อนอาจเป็นทางเลือก
การวินิจฉัย LCA ทำได้ทางคลินิก และจำเป็นต้องยืนยันด้วยการตรวจคลื่นไฟฟ้าจอตาและการยืนยันทางอณูพันธุศาสตร์โดยการตรวจยีน2) .
เมื่อพบการตอบสนองทางสายตาที่บกพร่องอย่างรุนแรงแต่กำเนิด (ขาดการจ้องและติดตาม) ให้สงสัย LCA ในทารกที่มีความบกพร่องทางการมองเห็น รุนแรงและสายตายาว มาก การค้นหา LCA โดยการตรวจทางอณูพันธุศาสตร์เป็นทางเลือกแรก4) .
การตรวจวินิจฉัยหลักแสดงไว้ด้านล่าง
การตรวจ ผลการตรวจ/ลักษณะ การตรวจคลื่นไฟฟ้าจอประสาทตา (ERG ) ไม่สามารถบันทึกได้หรือลดลงอย่างชัดเจนทั้งในที่มืดและสว่าง จำเป็นต้องทำ OCT (การถ่ายภาพตัดขวางด้วยแสง)จอประสาทตา ฝ่อ ชั้นนอกหายไป โซน ellipsoid หายไปFAF (การเรืองแสงอัตโนมัติของจอตา)แตกต่างกันตามชนิดย่อย (ชนิด RPE 65: หายไป, ชนิด GUCY2D: ปกติ) การตรวจทางพันธุกรรม (NGS ฯลฯ) จำเป็นสำหรับการวินิจฉัยที่แน่นอนและการระบุชนิดย่อย
การตรวจคลื่นไฟฟ้าจอประสาทตา : การตอบสนองของเซลล์รูปแท่ง และเซลล์รูปกรวย ทั้งสองชนิดหายไปหรือลดลงอย่างชัดเจน ERG ปกติจะตัดการวินิจฉัย LCA ออกไปOCT จอประสาทตา และโซน ellipsoid เกือบหายไป ในกรณีกลายพันธุ์ CRB1 จะพบความหนาของจอประสาทตา เพิ่มขึ้นอย่างขัดแย้ง (การเรียงชั้นหยาบ) 1) OCT แบบมือถือมีประโยชน์ในการตรวจทารกที่ตื่นอยู่หรือเด็กเล็กภายใต้การดมยาสลบ 4) FAF GUCY2D การเรืองแสงอัตโนมัติเป็นปกติ ในกรณีกลายพันธุ์ RPE 652) การตรวจทางพันธุกรรม : ใช้การหาลำดับรุ่นใหม่ (NGS), DNA microarray, การวิเคราะห์ความเชื่อมโยง อัตราการตรวจพบโดยรวมประมาณ 70-80% 2) ตั้งแต่ปี 2023 การตรวจแผงยีนสำหรับยีนก่อโรค IRD 82 ยีน (PrismGuide IRD Panel) ได้รับการครอบคลุมโดยประกัน และใช้กับผู้ป่วยที่เริ่มมีอาการเร็วซึ่งสงสัยว่าเป็น IRD ที่เกี่ยวข้องกับ RPE 65
การตรวจจอตา ในวัยทารก
ในวัยทารก จอตามักดูปกติ และความผิดปกติของจอตาจะปรากฏในภายหลัง ไม่มีรอยโรคจอประสาทตา ที่จำเพาะต่อ LCA หรือเฉพาะเจาะจงต่อชนิดย่อยใดๆ เนื่องจากความหลากหลายของฟีโนไทป์ทางคลินิกและการทับซ้อนทางพันธุกรรมกับโรคจอประสาทตา ทางพันธุกรรมอื่นๆ การตรวจทางพันธุกรรมจึงจำเป็นสำหรับการวินิจฉัยที่แน่นอน
จอประสาทตา เสื่อมชนิดสี (RP ) ที่เริ่มต้นเร็วการมองเห็น ส่วนกลางยังคงดี ไม่มีอาการตากระตุก จอประสาทตา เสื่อมชนิดรุนแรงที่เริ่มต้นเร็ว (EOSRD)การมองเห็น ลดลงอย่างรุนแรงหลังจากอายุ 1 ปี โดยไม่มีอาการตากระตุก 2) ตาบอดสีทั้งหมด (achromatopsia)อาการกลัวแสง และการเปลี่ยนแปลงเฉพาะของคลื่นไฟฟ้าจอตาตาบอดกลางคืนแต่กำเนิดชนิดคงที่ สายตาสั้น การมองเห็น ดีกว่า LCA และมีรูปแบบคลื่นไฟฟ้าจอตาเฉพาะกลุ่มอาการ Joubert : ร่วมกับภาวะสมองน้อย vermis ด้อยพัฒนา โรคไตเรื้อรัง และจอประสาทตา เสื่อมที่เริ่มต้นเร็ว
ยังไม่มีการรักษาที่เป็นรูปธรรมสำหรับ LCA ส่วนใหญ่ การจัดการในปัจจุบันมีดังนี้:
แก้ไขค่าสายตาผิดปกติ : ทำการแก้ไขค่าสายตาที่เหมาะสมสำหรับสายตายาว มาก เป็นต้น อาจมีค่าสายตาผิดปกติสูง ดังนั้นควรสั่งแว่นตาและพยายามฝึกการมองเห็น ฝึกการมองเห็น : ทำการฟื้นฟูสมรรถภาพทางการมองเห็น เพื่อใช้ประโยชน์สูงสุดจากการมองเห็น ที่เหลืออยู่ฝึกทดแทนการมองเห็น : เนื่องจากการมองเห็น บกพร่องอย่างรุนแรง ควรพิจารณาการสอนอักษรเบรลล์ การฝึกเดินด้วยไม้เท้าขาว และการใช้แว่นขยายอ่านหนังสือการดูแลผู้มีสายตาเลือนราง การให้คำปรึกษาทางพันธุกรรม การจัดการอาการกลัวแสง : แนะนำให้ใช้แว่นตาป้องกันแสงและลดการสัมผัสแสงการติดตามผลเป็นระยะ : ติดตามผลทางจักษุวิทยารวมถึงคลื่นไฟฟ้าจอตา ส่งต่อคลินิกผู้มีสายตาเลือนรางเมื่อจำเป็น
เมื่อได้รับการวินิจฉัยในวัยเด็ก ความวิตกกังวลของผู้ปกครองเกี่ยวกับอนาคตมีสูง ดังนั้นจึงจำเป็นต้องมีการดูแลครอบครัวโดยผู้เชี่ยวชาญ แม้ว่าปัจจุบันจะยังไม่มีวิธีการรักษาที่หายขาด แต่ความก้าวหน้าของการรักษาด้วยยีน การมองเห็น เทียม และการแพทย์ฟื้นฟูเป็นที่คาดหวัง และการให้ข้อมูลที่เหมาะสมและการสนับสนุนทางจิตใจแก่ผู้ป่วยและครอบครัวเป็นสิ่งสำคัญ
ในปี 2017 องค์การอาหารและยาสหรัฐอเมริกา (FDA) อนุมัติ voretigene neparvovec-rzyl (ชื่อการค้า Luxturna) สำหรับรักษา LCA2 ที่เกี่ยวข้องกับการกลายพันธุ์ของ RPE 65
สำเนาปกติของยีน RPE 65เยื่อบุผิวเม็ดสีจอประสาทตา โดยการฉีดใต้จอประสาทตา โดยใช้เวกเตอร์ไวรัสที่เกี่ยวข้องกับอะดีโนชนิดรีคอมบิแนนท์ (rAAV2) ขั้นตอนดำเนินการในห้องผ่าตัดน้ำวุ้นตา
การทดลองระยะที่ III 301 (Russell 2017)5) :
ผู้เข้าร่วม: ผู้ป่วย IRD ที่เกี่ยวข้องกับ RPE 65 จำนวน 31 ราย
ตัวชี้วัดหลัก: MLMT (การทดสอบการเคลื่อนไหวในสภาพแสงหลายระดับ; การทดสอบสังเกตพฤติกรรมในระดับความสว่างต่างๆ)
พบการปรับปรุงอย่างมีนัยสำคัญในเกณฑ์การกระตุ้นลานสายตาเต็มรูปแบบ (FST) เช่นกัน
ผลระยะยาวระยะที่ I/III (Maguire 2019)6) :
ความไวของจอประสาทตา การมองเห็น และประโยชน์เชิงหน้าที่ที่ถึงจุดสูงสุดที่ 6-12 เดือนหลังการรักษา มีแนวโน้มลดลงอย่างต่อเนื่องหลังจากนั้น
การเพิ่มขึ้นของความไวของจอประสาทตา สามารถช่วยให้ตาบอดกลางคืน และลานสายตาดีขึ้น
การทดลองระยะที่ III ในประเทศ (การทดลอง A11301) (Fujinami 2025)7) :
ผู้เข้าร่วม: ผู้ป่วยญี่ปุ่นที่มี IRD เกี่ยวข้องกับ RPE 65 จำนวน 4 ราย
เพิ่มความไวอย่างมีนัยสำคัญใน FST (เกณฑ์การกระตุ้นลานสายตาเต็มรูปแบบ) (เพิ่มความไวมากกว่า 10 เท่าถือว่ามีนัยสำคัญ)
ยืนยันการขยายลานสายตา ณ เวลา 1 ปีหลังการให้ยา
อาการไม่พึงประสงค์หลัก: ความผิดปกติของดวงตา รวมถึงอาการปวดตา (สันนิษฐานว่าเกี่ยวข้องกับขั้นตอนการให้ยา)
แนวทางการให้ยา:
ตาที่สองจะได้รับยาอย่างน้อย 6 วันหลังจากตาที่แรก
การกดภูมิคุ้มกัน: เริ่มให้สเตียรอยด์ 3 วันก่อนให้ยา และต่อเนื่องเป็นเวลา 14 วันหลังให้ยา
อย่างไรก็ตาม การกลายพันธุ์ของ RPE 65
การบำบัดด้วยยีน
การติดตามผลระยะยาวในระยะ I/III แสดงให้เห็นว่าประโยชน์ทางคลินิกซึ่งสูงสุดที่ 6–12 เดือนหลังการรักษาจะลดลงอย่างต่อเนื่อง 6) นอกจากนี้ ยังมีรายงานการฝ่อของคอรอยด์ และจอประสาทตา ส่วนรอบรอยบุ๋ม (perifoveal chorioretinal atrophy) เป็นภาวะแทรกซ้อนระยะยาว 8) เนื่องจากม่านตาอักเสบ ที่เกี่ยวข้องกับการบำบัดด้วยยีน (GTAU) อาจเกิดขึ้นได้ถึง 50% ของกรณีหลังผ่าตัด จึงจำเป็นต้องมีการกดภูมิคุ้มกันและการติดตามผลอย่างเหมาะสม 9)
Q
การบำบัดด้วยยีนสามารถใช้ได้กับผู้ป่วย LCA ทุกรายหรือไม่?
A
การบำบัดด้วยยีน ที่ได้รับการอนุมัติในปัจจุบัน (voretigene neparvovec / Luxturna®) มีข้อบ่งชี้เฉพาะสำหรับ LCA2 ที่เกิดจากการกลายพันธุ์แบบสองอัลลีลของ RPE 65RPE 65
Q
จะรับการบำบัดด้วยยีนในญี่ปุ่นได้อย่างไร?
A
เงื่อนไขคือต้องยืนยันการกลายพันธุ์แบบสองอัลลีลของ RPE 65 และมีเซลล์จอประสาทตา ที่มีชีวิตเพียงพอ ขั้นตอนแรกคือการวินิจฉัยทางพันธุกรรมโดยใช้ชุดตรวจยีน (PrismGuide IRD panel) แนะนำให้ส่งต่อผู้ป่วยไปยังสถานพยาบาลเฉพาะทางที่มีประสบการณ์ในการดูแลโรคจอประสาทตา ทางพันธุกรรม
พยาธิสรีรวิทยาของ LCA เกี่ยวข้องกับการหยุดชะงักของวงจรการมองเห็น (Visual Cycle) ซึ่งทำให้ดวงตาไม่สามารถส่งข้อมูลแสงได้
วงจรการมองเห็น เป็นชุดของปฏิกิริยาของเอนไซม์ระหว่างเยื่อบุผิวสีจอประสาทตา (RPE ) และจอประสาทตา ส่วนรับความรู้สึก โดยวิตามินเอจากอาหารจะถูกเมแทบอลิซึมเพื่อสร้าง 11-ซิส-เรตินัล (11-cis retinal) ซึ่งจำเป็นต่อการสร้างเม็ดสีที่ใช้ในการมองเห็น หากไม่มี 11-ซิส-เรตินัล จะไม่สามารถเริ่มต้นกระบวนการส่งสัญญาณแสง และสัญญาณประสาทการมองเห็น จะไม่ถูกส่งไปยังคอร์เทกซ์การมองเห็น การกลายพันธุ์ในยีนใดๆ ที่เข้ารหัสโปรตีนที่เกี่ยวข้องในปฏิกิริยาลูกโซ่นี้จะขัดขวางวงจรการมองเห็น และทำให้เกิดอาการของ LCA
ทางพยาธิวิทยาเนื้อเยื่อ มีหลักฐานการเกี่ยวข้องของจอประสาทตา ชั้นนอกและเซลล์รับแสง ซึ่งบ่งชี้ว่า LCA เป็นกระบวนการเสื่อม ไม่ใช่ภาวะเจริญผิดปกติ
GUCY2D (LCA1) : เข้ารหัส guanylate cyclase เฉพาะจอประสาทตา (GC-E) กระตุ้นการสังเคราะห์ cGMP ซึ่งเป็นกุญแจสำคัญในการถ่ายทอดสัญญาณแสง มีการระบุการกลายพันธุ์ที่เกี่ยวข้องกับโรคมากกว่า 140 ชนิด 88% ทำให้เกิด LCA แบบถ่ายทอดทาง autosomal recessive กรดอะมิโนตำแหน่ง 838 เป็นที่รู้จักในฐานะจุดร้อนของการกลายพันธุ์ 2) RPE 65 (LCA2)2) จำเป็นต่อการทำงานของทั้งเซลล์รูปแท่ง และเซลล์รูปกรวย การศึกษาล่าสุดชี้ให้เห็นถึงความเป็นไปได้ในการเกี่ยวข้องกับการ isomerization ของ lutein เป็น meso-zeaxanthin 2) การรักษาด้วยยีนชนิดเดียวที่ได้รับการอนุมัติCRB1 (LCA8) : มีความคล้ายคลึงกับโปรตีน crumbs ในแมลงหวี่ แสดงออกในส่วน inner segment ของเซลล์รับแสง และเซลล์ Müller มีความสำคัญต่อการรักษาสภาพขั้วของเซลล์ อยู่บนโครโมโซม 1q31.3 1) CEP290 (LCA10) : เกี่ยวข้องกับการทำงานของซิเลียของเซลล์รับแสง มีความถี่ของการกลายพันธุ์สูงที่สุดในบรรดายีนที่เกี่ยวข้องกับ LCA (ประมาณ 15%)NMNAT1 (LCA9) : เข้ารหัสเอนไซม์สำคัญในการสังเคราะห์ NAD (nicotinamide adenine dinucleotide) 2) LCA5 : เข้ารหัส lebercilin เกี่ยวข้องกับการทำงานของซิเลียและการขนส่งโปรตีนภายในซิเลีย 2) AIPL1 : ทำหน้าที่เป็น chaperone เฉพาะสำหรับ PDE6 (phosphodiesterase 6) การขาด AIPL1 ทำให้ PDE6 ไม่เสถียร → การเผาผลาญ cGMP ผิดปกติ → ความผิดปกติของช่องไอออน → การเสื่อมของเซลล์รับแสง 2)
ในการประเมินติดตามระยะยาวของ voretigene neparvovec (NCT 00481546, NCT 00643747) มีรายงานว่าพบจุดสูงสุดในช่วงแรกหลังการรักษา 6-12 เดือน ตามด้วยการลดลงอย่างต่อเนื่องของประโยชน์ทางคลินิก รวมถึงความไวของจอประสาทตา การมองเห็น และการทำงานที่ดีขึ้น 6) การศึกษา PERCEIVE (การศึกษาแบบไปข้างหน้า) รายงานข้อมูลความปลอดภัยและประสิทธิผล 2 ปีในทางปฏิบัติทางคลินิกจริง โดยพบม่านตาอักเสบ ที่เกี่ยวข้องกับการรักษาด้วยยีน (GTAU) ในผู้ป่วยมากถึง 50% 9)
พัฒนาโดย Editas Medicine เวกเตอร์ AAV5 บรรจุ Cas9 จาก S. aureus และ guide RNA สองตัว กำหนดเป้าหมายการกลายพันธุ์ในอินตรอนลึก (c.2991+1655A>G) ที่อยู่ในอินตรอน 26 ของ CEP290 3) การทดลอง first-in-human ยืนยันความปลอดภัยและแสดงให้เห็นถึงความทนทานที่ดีแม้ในขนาดที่ค่อนข้างสูง 3)
ในฐานะแนวทางที่ไม่ขึ้นกับยีน เทคนิคการแสดงออกของ channelrhodopsin ที่ตอบสนองต่อแสงบนเซลล์ประสาทจอประสาทตา ชั้นในที่เหลืออยู่กำลังถูกศึกษา 3) อาจใช้ได้กับ LCA ทุกประเภท (โดยไม่คำนึงถึงจีโนไทป์) และการทดลองทางคลินิกระยะแรกกำลังดำเนินอยู่
การบำบัดด้วยยีน สำหรับการกลายพันธุ์ GUCY2D และ AIPL1 กำลังดำเนินการในแบบจำลองสัตว์ และแสดงผลลัพธ์ที่มีแนวโน้มดีในการช่วยเหลือเซลล์รับแสง รูปแท่งและรูปกรวย
แนวทางของ LCA แบ่งออกเป็นสามรูปแบบ: คงที่ (ประมาณ 75%) แย่ลงเรื่อยๆ (ประมาณ 15%) และดีขึ้น (ประมาณ 10%) การกลายพันธุ์ AIPL1 สัมพันธ์กับการแย่ลงเรื่อยๆ ในขณะที่การกลายพันธุ์ RP GRIP1การมองเห็น เทียม การบำบัดด้วยยีน และการแพทย์ฟื้นฟู
Duan W, Zhou T, Jiang H, Zhang M, Hu M, Zhang L. A novel nonsense variant (c.1499C>G) in CRB1 caused Leber congenital amaurosis-8 in a Chinese family and a literature review. BMC Med Genomics. 2022;15(1):197. doi:10.1186/s12920-022-01356-z.
Mordà D, Alibrandi S, Scimone C, Rinaldi C, Scalinci SZ, Abate G, et al. Decoding pediatric inherited retinal dystrophies: Bridging genetic complexity and clinical heterogeneity. Progress in retinal and eye research. 2025;109:101405. doi:10.1016/j.preteyeres.2025.101405. PMID:40998214.
Botto C, Rucli M, Tekinsoy MD, Pulman J, Sahel JA, Dalkara D. Early and late stage gene therapy interventions for inherited retinal degenerations. Progress in retinal and eye research. 2022;86:100975. doi:10.1016/j.preteyeres.2021.100975. PMID:34058340.
Gurnani B, et al. Clinical approach to pediatric nystagmus: a comprehensive diagnostic algorithm. Clin Ophthalmol. 2025;19:1617-1636.
Russell S, Bennett J, Wellman JA, Chung DC, Yu ZF, Tillman A, et al. Efficacy and safety of voretigene neparvovec (AAV2-hRPE 65v2) in patients with RPE 65-mediated inherited retinal dystrophy: a randomised, controlled, open-label, phase 3 trial. Lancet (London, England). 2017;390(10097):849-860. doi:10.1016/S0140-6736(17)31868-8. PMID:28712537; PMCI D:PMC5726391.
Maguire AM, Russell S, Wellman JA, Chung DC, Yu ZF, Tillman A, et al. Efficacy, Safety, and Durability of Voretigene Neparvovec-rzyl in RPE 65 Mutation-Associated Inherited Retinal Dystrophy: Results of Phase 1 and 3 Trials. Ophthalmology. 2019;126(9):1273-1285. doi:10.1016/j.ophtha.2019.06.017. PMID:31443789.
Fujinami K, Akiyama K, Tsunoda K, et al. Efficacy and Safety of Voretigene Neparvovec in RPE 65-Retinopathy: Results of a Phase III Trial in Japan. Ophthalmol Sci. 2025;5:100876. doi:10.1016/j.xops.2025.100876. PMID:40910102; PMCI D:PMC12405627.
Gange WS, Sisk RA, Besirli CG, Lee TC, Havunjian M, Schwartz H, et al. Perifoveal Chorioretinal Atrophy after Subretinal Voretigene Neparvovec-rzyl for RPE 65-Mediated Leber Congenital Amaurosis. Ophthalmology. Retina. 2022;6(1):58-64. doi:10.1016/j.oret.2021.03.016. PMID:33838313; PMCI D:PMC8497635.
Fischer MD, Simonelli F, Sahni J, Holz FG, Maier R, Fasser C, et al. Real-World Safety and Effectiveness of Voretigene Neparvovec: Results up to 2 Years from the Prospective, Registry-Based PERCEIVE Study. Biomolecules. 2024;14(1). doi:10.3390/biom14010122. PMID:38254722; PMCI D:PMC10813228.