Stades 1 à 2
Stade 1 : présence uniquement de télangiectasies rétiniennes, sans exsudat.
Stade 2A : Télangiectasies avec exsudats extrafovéolaires.
Stade 2B : Exsudats atteignant la fovéa. Baisse d’acuité visuelle manifeste.

La maladie de Coats, décrite par George Coats en 1908, est une maladie vasculaire rétinienne idiopathique. Elle se caractérise par une dilatation anormale des capillaires rétiniens (télangiectasies) et une accumulation d’exsudats intra- et sous-rétiniens provenant des parois vasculaires.
La maladie est sporadique et non héréditaire, sans association avec des maladies systémiques ou des antécédents familiaux1). L’incidence est rare, de 0,09 pour 100 000 personnes2). Environ 75 % des patients sont des hommes, 95 % des cas sont unilatéraux, et elle survient principalement avant 20 ans (âge moyen d’environ 5 ans).
L’apparition chez l’adulte est très rare, mais présente un tableau clinique différent de la forme infantile. Les cas adultes sont moins sévères, avec une progression lente et une bonne réponse au traitement3). Dans une étude, seulement 21 % des 48 yeux adultes ont développé un décollement de rétine exsudatif, contre 81 % des cas infantiles3).
La forme légère est également appelée maladie des anévrismes miliaires de Leber et fait partie du spectre de la maladie de Coats. La télangiectasie maculaire idiopathique de type 1 est également considérée comme faisant partie du même spectre.
Des cas d’apparition après 35 ans ont été rapportés et sont reconnus comme maladie de Coats de l’adulte3). Par rapport à la forme infantile, la progression est plus lente, la fréquence de décollement exsudatif est plus faible, et la réponse au traitement est souvent bonne. Cependant, l’apparition est rare, et en cas de signes similaires chez l’adulte, un diagnostic différentiel avec d’autres maladies est important.
Lorsque la maladie survient pendant la petite enfance, l’enfant atteint exprime rarement des symptômes.
Dans les cas adultes, l’acuité visuelle est relativement préservée et la maladie peut être découverte fortuitement lors d’un examen de routine3).
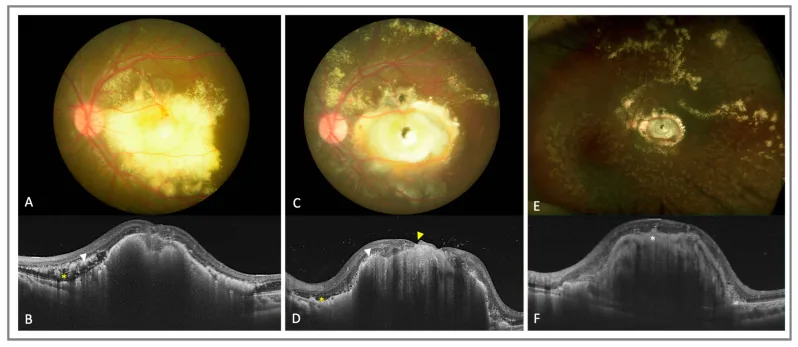
Les caractéristiques du fond d’œil sont des vaisseaux rétiniens anormalement dilatés concentrés dans la périphérie et une accumulation d’exsudats jaune-blanc sous-rétiniens et intrarétiniens. L’angiographie à la fluorescéine révèle une occlusion des vaisseaux rétiniens périphériques, des télangiectasies, des microanévrismes et des néovaisseaux. À un stade avancé, un décollement de rétine exsudatif se produit, pouvant aboutir à un décollement total. L’échographie, le scanner et l’IRM montrent un liquide sous-rétinien homogène dans tous les recoins, ce qui est un point de différenciation important avec le rétinoblastome, qui présente une image hétérogène avec calcifications.
Aux stades tardifs, des complications du segment antérieur telles que la rubéose irienne et le glaucome néovasculaire apparaissent. Le trou maculaire est une complication rare, rapportée dans seulement 7 cas dans la littérature4). Le nodule fibreux est un facteur de mauvais pronostic visuel, et l’OCTA confirme la présence de néovaisseaux de type 3 (SVC→DVC→complexe avasculaire) dans le nodule5).
La classification de Shields divise la maladie de Coats en 5 stades. Elle est utilisée pour choisir la stratégie thérapeutique et estimer le pronostic.
Stades 1 à 2
Stade 1 : présence uniquement de télangiectasies rétiniennes, sans exsudat.
Stade 2A : Télangiectasies avec exsudats extrafovéolaires.
Stade 2B : Exsudats atteignant la fovéa. Baisse d’acuité visuelle manifeste.
Stade 3
Stade 3A1 : Décollement rétinien subtotal extrafovéolaire.
Stade 3A2 : Décollement rétinien subtotal incluant la fovéa. Pronostic visuel défavorable.
Stade 3B : Décollement rétinien total. Intervention urgente nécessaire.
Stades 4 à 5
Stade 4 : Décollement rétinien total compliqué de glaucome secondaire. Peut être douloureux.
Stade 5 : Stade terminal. Atrophie du globe (phthisis). Énucléation envisagée.
Plusieurs maladies se présentent avec une leucocorie, la maladie de Coats n’en est qu’une. Le diagnostic différentiel le plus important est le rétinoblastome (Rb), qui nécessite une évaluation urgente en raison de son pronostic vital. D’autres diagnostics incluent la rétinopathie du prématuré, le syndrome de persistance du vitré primitif (PHPV) et l’endophtalmie. Voir la section « Diagnostic et méthodes d’examen » pour plus de détails.
La cause de la maladie de Coats est inconnue, sans association avec une maladie systémique ou des antécédents familiaux. Des rapports suggèrent une instabilité des chromosomes 3 et 13, ainsi qu’une association avec les gènes NDP (maladie de Norrie) et CRB1, mais cela n’est pas établi.
Le point de départ physiopathologique est la rupture de la barrière hémato-rétinienne interne (BHRi) 1). La diminution des péricytes (cellules de soutien de l’endothélium vasculaire) fragilise la paroi vasculaire, formant des capillaires anormalement dilatés et des anévrismes 1). La fuite et l’accumulation de composants plasmatiques dans la paroi vasculaire et les couches rétiniennes épaississent la paroi, entraînant un cercle vicieux d’exsudation supplémentaire 2).
Un environnement riche en VEGF favoriserait la dilatation des capillaires périphériques 2), ce qui constitue la base théorique du traitement anti-VEGF.
Le diagnostic repose sur une combinaison de plusieurs examens, et la distinction avec le rétinoblastome (Rb) est la priorité absolue.
L’examen du fond d’œil sous dilatation pupillaire révèle un réseau vasculaire anormal tortueux dans la périphérie et des exsudats sous-rétiniens jaune-blanc. Lorsque les exsudats atteignent la macula, ils prennent un aspect de plaque dure blanchâtre.
C’est l’un des examens les plus importants pour le diagnostic de la maladie de Coats. On observe une dilatation marquée des capillaires, des microanévrismes, des anastomoses artérioveineuses et des fuites de fluorescéine en forme d’ampoule (« light bulbs »). Ces signes sont essentiels pour délimiter la zone des vaisseaux anormaux et décider des sites de photocoagulation au laser.
Confirme l’absence de masse solide. Le Rb montre souvent des échos hyperéchogènes (calcifications) dans la masse solide en mode B, alors que la maladie de Coats ne forme pas de masse solide.
Évalue la présence de calcifications. Le Rb est fréquemment associé à des calcifications, contrairement à la maladie de Coats. Cette différence est un élément clé pour le diagnostic différentiel.
Les principaux points de différenciation entre la maladie de Coats et le rétinoblastome sont présentés ci-dessous.
| Élément | Maladie de Coats | Rétinoblastome |
|---|---|---|
| Âge de prédilection | Vers 5 ans | 1 à 2 ans |
| Sexe | Hommes 75 % | Aucune |
| Bilatéralité | Environ 5 % | Environ 40 % |
| Calcification | Absente | Présente (fréquente) |
| Tumeur solide | Absente | Présente |
| Échographie | Épanchement sous-rétinien | Tumeur solide, calcifications internes, ombre postérieure |
| IRM | Liquide sous-rétinien homogène | Hétérogène (signal tumoral) |
Les autres diagnostics différentiels incluent l’hémangiome rétinien, la maladie de von Hippel-Lindau, la persistance du vitré primitif hyperplasique (PHPV), la vitréorétinopathie exsudative familiale (FEVR), la toxocarose, la tumeur vasoproliférative, la maladie d’Eales, etc.
L’objectif du traitement est d’obstruer les vaisseaux anormaux et d’arrêter la production d’exsudats. Une approche progressive en fonction du stade est adoptée.
C’est le traitement de première intention. On coagule directement les vaisseaux anormalement dilatés et les microanévrismes identifiés par angiographie à la fluorescéine (FA), et on effectue également une photocoagulation dans les zones de non-perfusion périphériques. Chez l’enfant, l’intervention est réalisée sous anesthésie générale. Plusieurs séances sont souvent nécessaires, et après le traitement, une réévaluation régulière par FA et des coagulations supplémentaires sont répétées.
C’est l’option suivante pour les lésions de la périphérie antérieure ou les zones difficiles à photocoaguler. Elle peut être utilisée en combinaison avec la photocoagulation.
Photocoagulation au laser
Indications : Vaisseaux anormaux et lésions exsudatives des stades 1 à 3A.
Méthode : Coagulation directe des zones de télangiectasies et de non-perfusion sous guidage par FA. Chez l’enfant, sous anesthésie générale.
Caractéristiques : Peut être répété. La réévaluation régulière par FA après traitement et la coagulation supplémentaire font partie de la prise en charge standard.
Cryocoagulation
Indications : Lésions périphériques antérieures difficiles à photocoaguler, aide dans les cas graves de stade 3B ou moins.
Méthode : Application d’une sonde de cryocoagulation par voie transsclérale pour coaguler et occlure les vaisseaux anormaux.
Caractéristiques : Peut être réalisée même en milieu trouble ou dans les zones les plus périphériques.
Chirurgie vitréo-rétinienne
Indications : Stade 3B (décollement total de la rétine) ou plus, cas réfractaires à la cryocoagulation.
Méthode : Drainage externe ou vitrectomie pour drainer le liquide sous-rétinien et réappliquer la rétine par voie interne 8). Dans les cas graves, un drainage du liquide sous-rétinien combiné à une cerclage (scléral buckling) peut être réalisé.
Caractéristiques : Pour les cas compliqués de trou maculaire, la technique de retournement de la membrane limitante interne (ILM) a été rapportée comme efficace 4).
L’utilisation d’agents anti-VEGF pour la maladie de Coats n’a pas encore fait l’objet d’un consensus ; elle est considérée comme un traitement adjuvant en association avec la photocoagulation, et non comme un traitement standard.
Un cas de maladie de Coats chez l’adulte a été traité par une combinaison d’injection intravitréenne de ranibizumab 0,5 mg et de photocoagulation au laser, avec une amélioration de la meilleure acuité visuelle corrigée de compter les doigts à 20/603).
Dans un rapport sur des cas pédiatriques traités par injection intravitréenne de bevacizumab 1,25 mg, triamcinolone sous-ténonienne et laser combinés toutes les 6 semaines sous EUA (examen sous anesthésie générale), il a été noté qu’une exsudation paradoxale (rétinopathie exsudative paradoxale) peut survenir après le traitement2).
Des rapports récents indiquent que le brolucizumab a été efficace dans les cas résistants au bevacizumab2). Il a été suggéré que la thérapie anti-VEGF pourrait contribuer à prévenir la formation de nodules fibreux5).
L’énucléation est choisie en cas d’œil aveugle douloureux (stade 4 à 5) et lorsqu’il est difficile d’exclure un rétinoblastome.
Dans la maladie de Coats, des cas de récidive ou de redécollement peuvent survenir après plusieurs années, et il existe également des cas où l’apparition est bilatérale mais à des moments différents. Même après la fin du traitement, une réévaluation régulière par angiographie à la fluorescéine est nécessaire, et si de nouvelles lésions sont confirmées, une photocoagulation ou une cryocoagulation supplémentaire doit être réalisée.
À l’heure actuelle, il n’existe pas de consensus sur l’utilisation des anti-VEGF dans la maladie de Coats, et ils ne sont pas considérés comme un traitement standard. Des rapports s’accumulent sur leur utilisation en tant que thérapie adjuvante à la photocoagulation au laser, mais l’évaluation de leur efficacité et de leur sécurité nécessite des recherches supplémentaires.
Le mécanisme central de la maladie de Coats est la rupture de la barrière hémato-rétinienne interne (BHRi) 1).
La BHRi est constituée des cellules endothéliales des capillaires rétiniens et des péricytes qui les soutiennent. Dans la maladie de Coats, le nombre de péricytes est considérablement réduit, ce qui entraîne une diminution de la fonction de soutien endothélial vasculaire 1). Une diminution du nombre de cellules endothéliales elles-mêmes a également été confirmée par immunohistochimie et microscopie électronique 1).
La rupture de la BHR endothéliale entraîne une fuite et une accumulation de composants plasmatiques (principalement des lipoprotéines et du cholestérol) dans la paroi vasculaire, la rétine et l’espace sous-rétinien 1). Les lipides accumulés provoquent une infiltration de macrophages chargés de lipides (cellules spumeuses) et une réponse immunitaire granulomateuse, aggravant les lésions tissulaires 1).
Un environnement riche en VEGF favorise une dilatation supplémentaire des capillaires périphériques et contribue à la progression de la lésion 2). L’observation par OCTA a révélé la présence de néovaisseaux de type 3 (formés dans l’ordre SVC → DVC → complexe avasculaire) dans les nodules fibreux maculaires des lésions avancées 5), et la compréhension du processus de néovascularisation progresse.
Le mécanisme de formation du trou maculaire est considéré comme étant dû à un raccourcissement rétinien provoqué par la photocoagulation périphérique, qui génère une traction tangentielle et conduit à une perforation de la macula 4).
L’OCTA a permis d’évaluer de manière non invasive la structure microvasculaire à l’intérieur des nodules fibreux.
Ong et al. (2021) ont analysé en détail la structure vasculaire à l’intérieur des nodules maculaires à l’aide de l’OCTA et ont révélé la présence de néovaisseaux de type 3 formés dans l’ordre SVC → DVC → complexe avasculaire 5). Cette découverte est importante pour élucider le mécanisme de formation des nodules et comme cible pour la thérapie anti-VEGF.
Un phénomène d’aggravation paradoxale de l’exsudation après le début du traitement anti-VEGF a été rapporté, nécessitant l’élucidation du mécanisme et l’établissement de stratégies de prise en charge.
Kalavar et al. (2022) ont rapporté un cas de maladie de Coats chez un enfant ayant présenté une aggravation transitoire de l’exsudation et une formation d’exsudats maculaires en étoile (macular star formation) après le début du traitement2). Le brolucizumab s’est avéré efficace chez un patient résistant au bevacizumab, attirant l’attention en tant que nouvelle option thérapeutique2).
Nawrocka et al. (2023) ont rapporté un cas de vitrectomie utilisant la technique du flap inversé de la membrane limitante interne pour un trou maculaire associé à la maladie de Coats4). La fermeture du trou maculaire a été confirmée à 18 mois postopératoires, avec une meilleure acuité visuelle corrigée de 20/40. Le trou maculaire lié à la maladie de Coats est une complication rare, avec seulement environ 7 cas rapportés sur PubMed4).
Shields et al. (2019) ont analysé 351 yeux de patients atteints de la maladie de Coats sur 45 ans, montrant une amélioration des résultats thérapeutiques au fil des décennies9). Dalvin et al. (2019) ont analysé la même cohorte par catégorie d’âge, montrant que les cas d’apparition chez l’enfant ont tendance à être plus graves et à avoir un pronostic visuel plus défavorable que les cas d’apparition chez l’adulte10).
La maladie de Coats à début adulte est un concept pathologique historiquement sous-reconnu, mais le nombre de rapports augmente3). La clarification des différences cliniques avec la forme infantile (légère, progression lente, bonne réponse au traitement) progresse, et l’établissement de protocoles thérapeutiques appropriés chez l’adulte reste un défi.