Le rétinoblastome est une tumeur maligne de la rétine chez le nourrisson et le jeune enfant. Il résulte de la transformation maligne et de la prolifération des cellules rétiniennes immatures, et est considéré comme une maladie monogénique due à une mutation du gène RB1 situé sur le bras long du chromosome 13 (13q14.2). Il n’y a pas de prédominance de sexe, et 95 % des cas sont diagnostiqués avant l’âge de 5 ans.
L’incidence est de 1 pour 15 000 à 23 000 naissances, avec 70 à 80 nouveaux cas par an au Japon. Le rapport entre formes unilatérales et bilatérales est de 3:2, l’âge moyen au diagnostic étant de 21 mois pour les formes unilatérales et de 8 mois pour les bilatérales, ces dernières étant diagnostiquées plus précocement. Dans les pays développés, le taux de survie à 5 ans pour les formes intraoculaires localisées dépasse 95 %.
Le rétinoblastome est divisé en deux grandes catégories selon le type de mutation génétique.
Classification
Type de mutation
Caractéristiques pathologiques
Risque génétique
Héréditaire (mutation germinale)
Mutation RB1 dans la lignée germinale
Fréquence bilatérale et multifocale élevée
Transmis à 50 % des enfants
Non héréditaire (mutation somatique)
Mutation somatique dans une cellule rétinienne
Tumeur unilatérale et unique
Aucune transmission à la descendance
Héréditaire (mutation germinale) : Toutes les cellules du corps portent la première mutation. La seconde mutation conduit au cancer. Risque élevé de tumeurs bilatérales et multifocales, transmis à 1 enfant sur 2 (50 %). Risque de cancers secondaires comme l’ostéosarcome (15,7 % à 20 ans).
Non héréditaire (mutation somatique) : Les deux allèles du gène RB1 sont mutés dans une seule cellule rétinienne. Tumeur unilatérale et unique, sans risque de transmission à la descendance.
Cependant, même dans les cas unilatéraux, certaines mutations RB1 germinales sont incluses. Il ne faut pas nier le caractère héréditaire simplement parce qu’il est unilatéral, et il est nécessaire d’interpréter les antécédents familiaux, l’âge d’apparition et le nombre de tumeurs en se basant sur le conseil génétique et l’évaluation génétique. 1)
Classification par stade et taux de conservation du globe oculaire
La classification par stade est directement liée à la stratégie de traitement conservateur du globe oculaire.
Stade
État de la lésion
Taux estimé de conservation du globe oculaire
T1 (lésion intraoculaire précoce)
Limitée à l’œil, sans progression
Plus de 90 %
T2 (lésion intraoculaire avancée)
Progression intraoculaire
Environ 50 %
T3
Lésion avancée avec invasion extraoculaire
Environ 10 %
QLe rétinoblastome est-il héréditaire ?
A
Environ 40 % des cas sont héréditaires (mutation RB1 germinale) et se transmettent à l’enfant avec une probabilité de 50 %. Les 60 % restants sont non héréditaires (mutation somatique uniquement) et ne présentent aucun risque de transmission à la génération suivante. Les formes héréditaires ont tendance à être bilatérales et multifocales. Il est recommandé de réaliser un test génétique au moment du diagnostic et de bénéficier d’un conseil génétique.
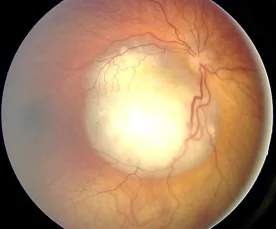
Aerts I, et al. Retinoblastoma. Orphanet J Rare Dis. 2006 Aug 25; 1:31. Figure 2. PMCID: PMC1586012. License: CC BY.
Image du fond d’œil d’un rétinoblastome observé comme une lésion blanche surélevée richement vascularisée, montrant l’aspect typique responsable de la leucocorie. Correspond aux signes du fond d’œil et à la leucocorie traités dans la section « 2. Principaux symptômes et signes cliniques ».
Dans de nombreux cas, la tumeur devient volumineuse à l’intérieur de l’œil et est découverte par une leucocorie. Lorsqu’elle se développe dans la macula, elle peut entraîner une mauvaise vision et un strabisme, ce qui permet sa découverte. Chez les enfants plus âgés, une baisse de l’acuité visuelle peut être ressentie ; chez les jeunes enfants, le fait de se frotter l’œil atteint peut être le premier symptôme.
Symptômes précoces
Leucocorie : symptôme initial le plus fréquent. La tumeur grossit dans l’œil et la pupille apparaît blanche.
Strabisme : dû à une mauvaise vision causée par une tumeur maculaire. L’œil malvoyant dévie vers l’extérieur.
Baisse de l’acuité visuelle : observée chez les enfants plus âgés.
Frottement de l’œil : observé chez les jeunes enfants ayant une mauvaise vision.
Symptômes à un stade avancé
Opacité cornéenne et augmentation de la pression intraoculaire : dues à la compression du cristallin par la tumeur ou à un glaucome néovasculaire (NVG).
Hyperémie conjonctivale et gonflement palpébral : signes inflammatoires.
Douleur : apparaît en raison de l’augmentation de la pression intraoculaire ou de la nécrose tumorale.
Exophtalmie : observée en cas d’extension extraoculaire.
On observe une lésion blanchâtre et surélevée riche en vaisseaux ; la présence de calcifications facilite le diagnostic définitif. Elle s’accompagne souvent de dissémination vitréenne (dispersion de cellules tumorales dans le vitré).
Test du reflet rouge (dépistage chez le nourrisson)
Le test du reflet rouge est un examen de base pour le dépistage des maladies oculaires chez le nourrisson. Il est normal si les pupilles des deux yeux sont de taille égale et présentent un reflet jaune-orangé vif et symétrique. Si le reflet est sombre ou trop brillant, ou s’il existe une différence entre les deux yeux, une investigation plus poussée est nécessaire.
QUne pupille blanche signifie-t-elle toujours un rétinoblastome ?
A
Les causes d’une pupille blanche incluent, outre le rétinoblastome, de nombreuses autres affections telles que la persistance du vitré primitif hyperplasique, la rétinopathie du prématuré, la maladie de Coats, etc. Cependant, en présence d’une pupille blanche, il est prioritaire de consulter rapidement un ophtalmologiste pour exclure un rétinoblastome. Tout retard de diagnostic a un impact direct sur le pronostic, donc une orientation immédiate est recommandée en cas de suspicion.
La cause est une mutation du gène RB1 situé sur le bras long du chromosome 13 (13q14.2). Le gène RB1 produit la protéine RB1 (protéine du rétinoblastome), qui joue un rôle important dans le contrôle de la division cellulaire.
Chaque cellule possède deux allèles du gène ; une mutation sur un seul allèle n’altère pas la fonction cellulaire, mais lorsque les deux allèles sont mutés, la cellule perd le contrôle de sa division et devient maligne (théorie des deux coups, hypothèse de Knudson).
Forme héréditaire (mutation germinale) : le premier coup est une mutation germinale (présente dans toutes les cellules). Le second coup survient dans une cellule somatique, conduisant à la cancérisation. Elle prédispose aux tumeurs bilatérales et multiples.
Forme non héréditaire (mutation somatique) : les premier et second coups surviennent tous deux dans une même cellule somatique. Elle se présente sous forme de tumeur unilatérale et unique.
Les antécédents familiaux sont le facteur de risque le plus important. La définition du risque selon les recommandations de l’AAOOP (American Association of Ophthalmic Oncologists and Pathologists) est présentée ci-dessous1).
Catégorie de risque
Définition
Valeur du risque
Élevé
Parent atteint de Rb bilatéral, ou porteur d’une mutation germinale RB1 chez un parent au 1er ou 2e degré
Dans les cas héréditaires, il faut être attentif au risque de second cancer. L’ostéosarcome est typique, survenant chez 15,7 % des cas héréditaires en 20 ans. Les seconds cancers apparaissent souvent après l’âge de 10 ans.
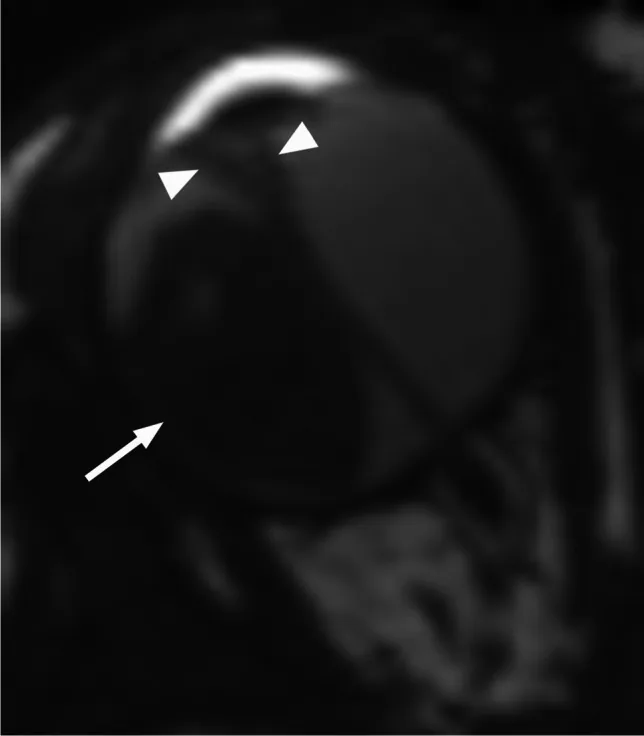
Rumboldt Z, Dodig D, Galluzzi P, et al. Retinoblastoma and beyond: pediatric orbital mass lesions. Neuroradiology. 2025;67(2):469-492. Figure 2. PMID: 39729290; PMCID: PMC11893699; DOI: 10.1007/s00234-024-03517-6. License: CC BY.
IRM pondérée T2 en coupe axiale montrant un rétinoblastome exophytique (flèche) avec un décollement de rétine secondaire en forme de V (tête de flèche). Correspond aux schémas de croissance exophytique et endophytique et aux résultats IRM traités dans la section « 4. Diagnostic et méthodes d’examen ».
La biopsie d’une tumeur intraoculaire n’est pas réalisée. Les lésions intraoculaires peuvent être observées directement à travers les tissus transparents, et la précision du diagnostic clinique est élevée. De plus, la biopsie d’une tumeur intraoculaire peut entraîner une dissémination extraoculaire des cellules tumorales, ce qui expose à un risque de métastases inévitable. En cas de traitement conservateur de l’œil, le traitement est débuté sur la base du diagnostic clinique.
Examen du fond d’œil (principal) : lésion blanche surélevée riche en vaisseaux + calcification pour un diagnostic clinique définitif
Échographie : confirmation d’une tumeur solide et de calcifications. Attention : la calcification est rare chez les enfants de plus de 5 ans.
IRM : signal modéré en T1, légèrement hypo-intense en T2, rehaussement après contraste. Essentielle pour évaluer l’infiltration du nerf optique et l’extension extraoculaire.
IRM cérébrale : environ 3 % des cas bilatéraux développent un rétinoblastome trilateral (tumeur pinéale), donc un dépistage est obligatoire.
Scanner : excellent pour visualiser les calcifications mais exposition aux radiations. Si l’IRM est réalisable, le scanner est auxiliaire.
Le diagnostic différentiel des maladies provoquant une leucocorie est primordial.
Persistance du vitré primitif (hyperplasie du vitré primitif) : L’échographie permet de vérifier la présence ou non d’une tumeur solide
Rétinopathie du prématuré : Les antécédents de prématurité et de faible poids de naissance sont des indices pour le diagnostic différentiel
Maladie de Coats : Accumulation d’exsudat jaune-blanc sous la rétine. Le schéma vasculaire tumoral est différent
Hamartome astrocytaire : Le diagnostic différentiel repose sur la présence de vaisseaux tumoraux, la localisation en OCT et l’absence de croissance
Cysticercose intraoculaire : Rare, mais peut imiter le rétinoblastome. Un cas rapporté chez un garçon de 4 ans avec leucocorie, où l’énucléation pour suspicion de Rb a révélé une cysticercose à l’examen pathologique3)
Pour le dépistage des enfants ayant des antécédents familiaux de Rb, la recommandation AAOOP 2018 est largement utilisée à l’échelle internationale1).
Risque
Calendrier de dépistage
Âge de fin
Élevé (>7,5%)
Naissance à 8 sem : toutes les 2 à 4 sem → 8 à 12 sem : 1 fois/mois → 1 à 2 ans : tous les 2 mois → 2 à 3 ans : tous les 3 mois → 3 à 4 ans : tous les 4 mois → 4 à 7 ans : tous les 6 mois
7 ans (porteurs de mutation RB1 : à vie)
Intermédiaire (1 à 7,5%)
Naissance à 3 mois : 1 fois/mois → diminution progressive
7 ans
Faible (<1%)
Naissance à 3 mois : 1 fois/mois → diminution progressive
7 ans
Une étude de cohorte rétrospective nationale néerlandaise (1991-2019, 38 cas familiaux sur 332) a montré que les 28 patients ayant bénéficié d’un dépistage complet ont tous été diagnostiqués avant l’âge d’un an (médiane 18 jours), tandis que chez les 10 patients avec un dépistage incomplet, le diagnostic a été considérablement retardé (médiane 420 jours, intervalle 59 jours à 4,8 ans)2). Une révision du protocole visant à réduire l’âge de fin de dépistage à 2 ans pour les groupes à faible risque (<3%) a également été proposée2).
Dans les cas familiaux, la poursuite du dépistage du fond d’œil dès la naissance a un impact direct sur le pronostic. Les études de registre classiques montrent également que le moment du diagnostic dans les cas familiaux est étroitement lié à la fréquence du dépistage, et on s’oriente actuellement vers une individualisation de la date de fin du dépistage en fonction de la présence ou non d’une mutation RB1.1, 2)
QSi un membre de la famille a des antécédents de rétinoblastome, jusqu'à quand l'enfant doit-il être suivi ?
A
L’AAOOP recommande un examen du fond d’œil régulier jusqu’à l’âge de 7 ans. En cas de dépistage complet, le diagnostic est généralement posé avant l’âge d’un an. Si le test génétique exclut un risque de mutation RB1, le dépistage peut être arrêté plus tôt. Pour les porteurs d’une mutation RB1, un suivi irrégulier tous les 1 à 2 ans après l’âge de 7 ans est recommandé.
Pour les lésions intraoculaires précoces avec un potentiel de vision conservé, un traitement conservateur de l’œil est activement entrepris. Aux stades intraoculaires avancés, la fonction visuelle est souvent compromise, mais un traitement conservateur peut être envisagé si la famille le souhaite. Le traitement nécessite une expertise hautement spécialisée, et une orientation précoce vers un centre spécialisé est cruciale.
Indiqué pour les tumeurs jusqu’à environ 3 mm de diamètre. L’irradiation directe au laser infrarouge permet un contrôle local d’environ 90 %. En cas de tumeur maculaire, une chimiothérapie systémique préalable est recommandée pour éviter des lésions visuelles irréversibles.
② Cryothérapie
Indiquée pour les tumeurs d’environ 3 mm situées en périphérie de l’équateur. La technique de triple congélation-décongélation (triple freeze-thaw) est courante et permet un contrôle local d’environ 90 %, similaire au laser.
③ Curiethérapie
Elle est indiquée pour les tumeurs localisées, éloignées de la papille optique, d’une épaisseur ≤ 5 mm et d’un diamètre ≤ 15 mm. Au Japon et en Europe, on utilise le ¹⁰⁶Ru (ruthenium-106, source β), tandis qu’en Amérique du Nord, on utilise une source d’¹²⁵I. Le traitement consiste à suturer temporairement la source sur la sclère en regard de la tumeur, nécessitant une salle de traitement spéciale, ce qui limite les établissements. Un contrôle local de 80 à 90 % est possible.
④ Chimiothérapie systémique (protocole VEC)
Elle est le traitement de première intention pour les tumeurs intraoculaires à un stade avancé. Une polychimiothérapie associant trois agents est largement utilisée, mais le taux de guérison par ce seul traitement est inférieur à 10 %. Après réduction tumorale, un traitement local (laser, cryothérapie, curiethérapie) est réalisé pour consolider.
Médicament
Dose (basée sur la surface corporelle)
Dose (basée sur le poids pour ≤ 36 mois)
Calendrier d’administration
Vincristine (Oncovin®)
1,5 mg/m²
0,05 mg/kg
Jour 1
Carboplatine (Paraplatine®)
560 mg/m²
18,6 mg/kg
Jour 1
Étoposide (Vepesid®)
150 mg/m²
5 mg/kg
jour 1, 2
Répéter 2 à 6 fois toutes les 3 à 4 semaines (tous en perfusion intraveineuse).
Administration directe d’un médicament (melphalan ; Alkeran® solution injectable) dans l’artère ophtalmique à l’aide d’un cathéter. Cela permet d’administrer une dose élevée localement dans l’œil tout en réduisant la dose systémique, diminuant ainsi les effets secondaires comme la myélosuppression. Bien que non couvert par l’assurance maladie, il s’agit d’un traitement expérimental pratiqué dans plus de 20 pays.
Pour les disséminations vitréennes, l’efficacité de la chimiothérapie systémique et de l’injection artérielle est limitée, donc on associe une injection intravitréenne de melphalan (Alkeran® solution injectable). Il s’agit d’un traitement expérimental non couvert par l’assurance maladie.
⑦ Radiothérapie externe
Irradiation fractionnée par rayons X de 40 à 46 Gy. Jusqu’aux années 1990, c’était le pilier du traitement conservateur de l’œil, mais la déformation orbitaire et l’augmentation des cancers secondaires sont devenues évidentes, et elle n’est plus utilisée que lorsque les autres traitements ne permettent pas le contrôle.
La positivité de la marge du nerf optique et l’infiltration extrasclérale sont des indications absolues de traitement adjuvant, comprenant une chimiothérapie systémique et une radiothérapie. L’infiltration choroïdienne marquée et l’infiltration du nerf optique au-delà de la lame criblée sont évaluées comme facteurs de risque relatifs de métastase.
Infiltration de la chambre antérieure ou de l’iris : risque de dissémination extraoculaire
Lorsqu’aucune récupération de la fonction visuelle n’est attendue : priorité au pronostic vital
QEst-il possible de conserver l'œil ?
A
Pour les lésions intraoculaires précoces (T1), la conservation de l’œil est possible dans plus de 90 % des cas. Une chimiothérapie systémique (VEC) réduit la tumeur, suivie d’un traitement de consolidation par laser, cryothérapie ou curiethérapie. Dans les cas avancés (T3), le taux de conservation oculaire est d’environ 10 %, et l’énucléation peut être nécessaire. Le choix du traitement est déterminé par un spécialiste en fonction du stade et des perspectives de fonction visuelle.
Le gène RB1 situé sur le bras long du chromosome 13 (13q14.2) code pour la protéine RB1 (pRb), qui joue un rôle clé dans le contrôle de la division cellulaire. pRb se lie au facteur de transcription E2F et inhibe la transition G1/S du cycle cellulaire, agissant ainsi comme une protéine suppresseur de tumeur régulant la prolifération cellulaire.
Selon la théorie des deux coups proposée par Knudson, la malignité survient lorsque les deux allèles du gène RB1 dans une cellule sont inactivés.
Héréditaire : Le premier coup (mutation germinale) est présent dans toutes les cellules. Lorsqu’un deuxième coup (mutation somatique, perte d’hétérozygotie, etc.) survient dans une cellule rétinienne, un cancer se développe. Ainsi, les tumeurs bilatérales et multifocales sont fréquentes, et le diagnostic est généralement plus précoce.
Non héréditaire : Le premier et le deuxième coup surviennent tous deux sous forme de mutations somatiques dans la même cellule rétinienne. La probabilité que les deux coups se produisent par hasard dans une même cellule est faible, donc les tumeurs sont souvent unilatérales et uniques, et le diagnostic tend à être plus tardif que dans les cas héréditaires.
Dans les cas héréditaires, le premier coup de RB1 est présent dans toutes les cellules du corps. Si un deuxième coup survient dans des cellules autres que la rétine (os, tissus mous, etc.), une tumeur maligne secondaire primitive se développe. L’ostéosarcome est le plus fréquent, survenant souvent après l’âge de 10 ans. Chez les patients héréditaires ayant reçu une radiothérapie externe, le risque de cancer secondaire est encore accru, de sorte que la radiothérapie externe est désormais utilisée de manière limitée.
L’IAC (chimiothérapie intra-artérielle) consiste à injecter du melphalan directement dans l’artère ophtalmique, permettant une administration de médicament à haute concentration dans l’œil tout en minimisant la toxicité systémique. Elle peut élargir les possibilités de conservation du globe oculaire même dans les cas avancés avec dissémination vitréenne. Actuellement, une évaluation à long terme des résultats dans de grandes cohortes est en cours.
L’injection intravitréenne de melphalan est réalisée pour traiter la dissémination vitréenne, mais la standardisation internationale des protocoles de dose et d’intervalle d’administration reste un défi. Plusieurs séries de cas ont rapporté des taux élevés de contrôle de la dissémination, et des essais prospectifs futurs devraient établir sa place.
Dans une étude de cohorte néerlandaise, une corrélation claire a été démontrée entre la participation à un dépistage complet et un diagnostic précoce (médiane de 18 jours), et les effets néfastes de l’interruption du dépistage ont été quantifiés 2). De plus, des propositions visant à réduire l’âge de fin du dépistage (2 ans) pour les groupes à faible risque font progresser l’optimisation des protocoles basée sur la stratification des risques 2). La diffusion mondiale des recommandations de l’AAOOP et la standardisation des protocoles régionaux sont des défis futurs 1).
Association avec des malformations cérébrales congénitales
Bien que rares, des cas de rétinoblastome associé à des malformations cérébrales congénitales ou à des anomalies chromosomiques ont été rapportés. Un rapport de rétinoblastome bilatéral avec syndrome de Dandy-Walker souligne l’importance d’une évaluation systémique incluant le contexte neurodéveloppemental en plus des symptômes oculaires. 4)
Avec la diffusion du séquençage de nouvelle génération (NGS), la sensibilité de détection des mutations germinales de RB1 s’est améliorée. La possibilité d’une surveillance par biopsie liquide (ADN tumoral circulant) est également étudiée, et son application à la stratification pronostique sans biopsie invasive est attendue.
Optimisation de la surveillance des cancers secondaires
La standardisation des protocoles de surveillance des cancers secondaires pour les survivants à long terme du rétinoblastome héréditaire est nécessaire. En particulier, la fréquence et l’âge de fin du dépistage multi-organes par IRM corps entier font l’objet d’une accumulation continue de preuves par des études de cohorte.
Positionnement international de l’IAC et de l’IVitC
L’injection artérielle oculaire sélective (IAC) et la chimiothérapie intravitréenne (IVitC) s’imposent internationalement comme des traitements qui ont amélioré les taux de conservation du globe oculaire dans les cas avancés ou avec dissémination vitréenne. Dans les revues sur les traitements conservateurs, elles sont positionnées comme des stratégies élargissant la conservation du globe sans augmenter le risque métastatique, et les résultats sont influencés par l’état d’adoption dans chaque pays et la centralisation dans des centres spécialisés. 5)
Disparités thérapeutiques dans les pays en développement
Alors que le taux de survie à 5 ans dans les pays développés dépasse 95 %, il serait de seulement 25 à 70 % dans les pays à revenu faible ou intermédiaire d’Afrique et d’Asie. Les revues systématiques et les analyses par niveau de revenu national montrent systématiquement des disparités dans les taux de survie et de conservation du globe oculaire, les principaux facteurs étant un diagnostic tardif, un accès insuffisant aux soins et un manque d’installations spécialisées. La diffusion de programmes de dépistage communautaires, y compris la méthode du réflexe rouge, est un défi international. 6, 7)
Skalet AH, Gombos DS, Gallie BL, Kim JW, Shields CL, Marr BP, Plon SE, Chévez-Barrios P. Screening Children at Risk for Retinoblastoma: Consensus Report from the American Association of Ophthalmic Oncologists and Pathologists. Ophthalmology. 2018;125(3):453-458. doi:10.1016/j.ophtha.2017.09.001. PMID:29056300.
Badalova NA, van Hoefen Wijsard M, Dommering CJ, et al. At what age could screening for familial retinoblastoma be stopped? Revised Dutch retrospective population-based cohort study 1991-2019. Ophthalmology. 2024;131(10):1189-1196.
Jakati S, Vempuluru VS, Kaliki S. Intraocular Cysticercosis Masquerading as Cavitary Retinoblastoma. Ophthalmology. 2025;132(4):e68. doi:10.1016/j.ophtha.2024.05.017. PMID:38878044.
Lomi N, Das D, Chawla B, Parampalli Ravindra A. Retinoblastoma in Dandy-Walker Syndrome. Cureus. 2025;17(8):e89663. doi:10.7759/cureus.89663. PMID:40926918; PMCID:PMC12415497.
Francis L. Munier, Maja Beck-Popovic, Guillermo L. Chantada, David Cobrinik, Tero T. Kivelä, Dietmar Lohmann, Philippe Maeder, Annette C. Moll, et al. Conservative management of retinoblastoma: Challenging orthodoxy without compromising the state of metastatic grace. “Alive, with good vision and no comorbidity”. Progress in Retinal and Eye Research. 2019;73:100764. doi:10.1016/j.preteyeres.2019.05.005.
Wong ES, Choy RW, Zhang Y, et al. Global retinoblastoma survival and globe preservation: a systematic review and meta-analysis. Lancet Glob Health. 2022;10(3):e380-e389.
Global Retinoblastoma Study Group, Fabian ID, Abdallah E, Abdullahi SU, Abdulqader RA, Adamou Boubacar S, Ademola-Popoola DS, Adio A, Afshar AR, Aggarwal P, Aghaji AE, Ahmad A, Akib MNR, Al Harby L, Al Ani MH, Alakbarova A, Portabella SA, Al-Badri SAF, Alcasabas APA, Al-Dahmash SA, Alejos A, Alemany-Rubio E, Alfa Bio AI, Alfonso Carreras Y, Al-Haddad C, Al-Hussaini HHY, Ali AM, Alia DB, Al-Jadiry MF, Al-Jumaily U, Alkatan HM, All-Eriksson C, Al-Mafrachi AARM, Almeida AA, Alsawidi KM, Al-Shaheen AASM, Al-Shammary EH, Amiruddin PO, Antonino R, Astbury NJ, Atalay HT, Atchaneeyasakul LO, Atsiaya R, Attaseth T, Aung TH, Ayala S, Baizakova B, Balaguer J, Balayeva R, Balwierz W, Barranco H, Bascaran C, Beck Popovic M, Benavides R, Benmiloud S, Bennani Guebessi N, Berete RC, Berry JL, Bhaduri A, Bhat S, Biddulph SJ, Biewald EM, Bobrova N, Boehme M, Boldt HC, Bonanomi MTBC, Bornfeld N, Bouda GC, Bouguila H, Boumedane A, Brennan RC, Brichard BG, Buaboonnam J, Calderón-Sotelo P, Calle Jara DA, Camuglia JE, Cano MR, Capra M, Cassoux N, Castela G, Castillo L, Català-Mora J, Chantada GL, Chaudhry S, Chaugule SS, Chauhan A, Chawla B, Chernodrinska VS, Chiwanga FS, Chuluunbat T, Cieslik K, Cockcroft RL, Comsa C, Correa ZM, Correa Llano MG, Corson TW, Cowan-Lyn KE, Csóka M, Cui X, Da Gama IV, Dangboon W, Das A, Das S, Davanzo JM, Davidson A, De Potter P, Delgado KQ, Demirci H, Desjardins L, Diaz Coronado RY, Dimaras H, Dodgshun AJ, Donaldson C, Donato Macedo CR, Dragomir MD, Du Y, Du Bruyn M, Edison KS, Eka Sutyawan IW, El Kettani A, Elbahi AM, Elder JE, Elgalaly D, Elhaddad AM, Elhassan MMA, Elzembely MM, Essuman VA, Evina TGA, Fadoo Z, Fandiño AC, Faranoush M, Fasina O, Fernández DDPG, Fernández-Teijeiro A, Foster A, Frenkel S, Fu LD, Fuentes-Alabi SL, Gallie BL, Gandiwa M, Garcia JL, García Aldana D, Gassant PY, Geel JA, Ghassemi F, Girón AV, Gizachew Z, Goenz MA, Gold AS, Goldberg-Lavid M, Gole GA, Gomel N, Gonzalez E, Gonzalez Perez G, González-Rodríguez L, Garcia Pacheco HN, Graells J, Green L, Gregersen PA, Grigorovski NDAK, Guedenon KM, Gunasekera DS, Gündüz AK, Gupta H, Gupta S, Hadjistilianou T, Hamel P, Hamid SA, Hamzah N, Hansen ED, Harbour JW, Hartnett ME, Hasanreisoglu M, Hassan S, Hassan S, Hederova S, Hernandez J, Hernandez LMC, Hessissen L, Hordofa DF, Huang LC, Hubbard GB, Hummlen M, Husakova K, Hussein Al-Janabi AN, Ida R, Ilic VR, Jairaj V, Jeeva I, Jenkinson H, Ji X, Jo DH, Johnson KP, Johnson WJ, Jones MM, Kabesha TBA, Kabore RL, Kaliki S, Kalinaki A, Kantar M, Kao LY, Kardava T, Kebudi R, Kepak T, Keren-Froim N, Khan ZJ, Khaqan HA, Khauv P, Kheir WJ, Khetan V, Khodabande A, Khotenashvili Z, Kim JW, Kim JH, Kiratli H, Kivelä TT, Klett A, Komba Palet JEK, Krivaitiene D, Kruger M, Kulvichit K, Kuntorini MW, Kyara A, Lachmann ES, Lam CPS, Lam GC, Larson SA, Latinovic S, Laurenti KD, Le BHA, Lecuona K, Leverant AA, Li C, Limbu B, Long QB, López JP, Lukamba RM, Lumbroso L, Luna-Fineman S, Lutfi D, Lysytsia L, Magrath GN, Mahajan A, Majeed AR, Maka E, Makan M, Makimbetov EK, Manda C, Martín Begue N, Mason L, Mason JO, Matende IO, Materin M, Mattosinho CCDS, Matua M, Mayet I, Mbumba FB, McKenzie JD, Medina-Sanson A, Mehrvar A, Mengesha AA, Menon V, Mercado GJVD, Mets MB, Midena E, Mishra DKC, Mndeme FG, Mohamedani AA, Mohammad MT, Moll AC, Montero MM, Morales RA, Moreira C, Mruthyunjaya P, Msina MS, Msukwa G, Mudaliar SS, Muma KI, Munier FL, Murgoi G, Murray TG, Musa KO, Mushtaq A, Mustak H, Muyen OM, Naidu G, Nair AG, Naumenko L, Ndoye Roth PA, Nency YM, Neroev V, Ngo H, Nieves RM, Nikitovic M, Nkanga ED, Nkumbe H, Nuruddin M, Nyaywa M, Obono-Obiang G, Oguego NC, Olechowski A, Oliver SCN, Osei-Bonsu P, Ossandon D, Paez-Escamilla MA, Pagarra H, Painter SL, Paintsil V, Paiva L, Pal BP, Palanivelu MS, Papyan R, Parrozzani R, Parulekar M, Pascual Morales CR, Paton KE, Pawinska-Wasikowska K, Pe’er J, Peña A, Peric S, Pham CTM, Philbert R, Plager DA, Pochop P, Polania RA, Polyakov VG, Pompe MT, Pons JJ, Prat D, Prom V, Purwanto I, Qadir AO, Qayyum S, Qian J, Rahman A, Rahman S, Rahmat J, Rajkarnikar P, Ramanjulu R, Ramasubramanian A, Ramirez-Ortiz MA, Raobela L, Rashid R, Reddy MA, Reich E, Renner LA, Reynders D, Ribadu D, Riheia MM, Ritter-Sovinz P, Rojanaporn D, Romero L, Roy SR, Saab RH, Saakyan S, Sabhan AH, Sagoo MS, Said AMA, Saiju R, Salas B, San Román Pacheco S, Sánchez GL, Sayalith P, Scanlan TA, Schefler AC, Schoeman J, Sedaghat A, Seregard S, Seth R, Shah AS, Shakoor SA, Sharma MK, Sherief ST, Shetye NG, Shields CL, Siddiqui SN, Sidi Cheikh S, Silva S, Singh AD, Singh N, Singh U, Singha P, Sitorus RS, Skalet AH, Soebagjo HD, Sorochynska T, Ssali G, Stacey AW, Staffieri SE, Stahl ED, Stathopoulos C, Stirn Kranjc B, Stones DK, Strahlendorf C, Suarez MEC, Sultana S, Sun X, Sundy M, Superstein R, Supriyadi E, Surukrattanaskul S, Suzuki S, Svojgr K, Sylla F, Tamamyan G, Tan D, Tandili A, Tarrillo Leiva FF, Tashvighi M, Tateshi B, Tehuteru ES, Teixeira LF, Teh KH, Theophile T, Toledano H, Trang DL, Traoré F, Trichaiyaporn S, Tuncer S, Tyau-Tyau H, Umar AB, Unal E, Uner OE, Urbak SF, Ushakova TL, Usmanov RH, Valeina S, van Hoefen Wijsard M, Varadisai A, Vasquez L, Vaughan LO, Veleva-Krasteva NV, Verma N, Victor AA, Viksnins M, Villacís Chafla EG, Vishnevskia-Dai V, Vora T, Wachtel AE, Wackernagel W, Waddell K, Wade PD, Wali AH, Wang YZ, Weiss A, Wilson MW, Wime ADC, Wiwatwongwana A, Wiwatwongwana D, Wolley Dod C, Wongwai P, Xiang D, Xiao Y, Yam JC, Yang H, Yanga JM, Yaqub MA, Yarovaya VA, Yarovoy AA, Ye H, Yousef YA, Yuliawati P, Zapata López AM, Zein E, Zhang C, Zhang Y, Zhao J, Zheng X, Zhilyaeva K, Zia N, Ziko OAO, Zondervan M, Bowman R.. Global Retinoblastoma Presentation and Analysis by National Income Level. JAMA Oncol. 2020;6(5):685-695. doi:10.1001/jamaoncol.2019.6716. PMID:32105305; PMCID:PMC7047856.
Copiez le texte de l'article et collez-le dans l'assistant IA de votre choix.
Article copié dans le presse-papiers
Ouvrez un assistant IA ci-dessous et collez le texte copié dans la conversation.