L’hémangiome capillaire rétinien (retinal capillary hemangioma) est une tumeur bénigne orange qui survient sur la rétine ou la papille optique des jeunes. En raison de sa similarité pathologique avec l’hémangioblastome cérébral, il est également appelé hémangioblastome rétinien (retinal hemangioblastoma) ces dernières années. Il peut être unique/multiple, unilatéral/bilatéral, sporadique/syndromique.
Lorsque seul un hémangioblastome rétinien survient de manière sporadique, on parle de maladie de Von Hippel. Lorsqu’il est associé à un syndrome tumoral systémique, on diagnostique une maladie de VHL (von Hippel-Lindau). Dans le glossaire de la Société japonaise d’ophtalmologie, conformément aux directives de pratique clinique pour VHL (édition 2024), le terme « hémangiome rétinien » est utilisé, mais dans cet article, nous utilisons le terme courant « hémangiome capillaire rétinien ».
La maladie de VHL est un syndrome de tumeurs héréditaires autosomique dominant causé par une mutation du gène suppresseur de tumeur VHL (3p25-26). Sa fréquence est estimée à 1 personne sur 36 000. Dans la maladie de VHL, en plus de l’hémangiome rétinien, des lésions surviennent dans de multiples organes : hémangioblastomes du cervelet, du bulbe rachidien, du pont et de la moelle épinière, carcinome rénal, phéochromocytome, kystes des organes abdominaux (pancréas, reins, glandes surrénales), etc. Le pronostic vital dépend également de la gestion des lésions systémiques, d’où la nécessité d’une collaboration pluridisciplinaire.
QDans quels cas suspecte-t-on une maladie de VHL ?
A
Selon les critères diagnostiques des directives de pratique clinique pour VHL (édition 2024), en présence d’antécédents familiaux, un diagnostic est posé avec une ou plusieurs lésions (hémangioblastome, hémangiome rétinien, etc.) 1). En l’absence d’antécédents familiaux, le diagnostic nécessite au moins deux lésions (incluant un hémangioblastome ou un hémangiome rétinien), ou la confirmation d’une mutation du gène VHL plus une lésion. Chez les jeunes présentant un hémangiome rétinien sporadique, en particulier avant l’âge de 10 ans, un diagnostic ultérieur de maladie de VHL est fréquent, il faut donc envisager activement un bilan systémique et un test génétique.
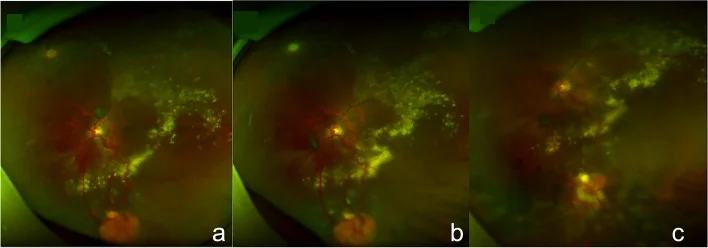
Guo J, Du L, Zhou P, et al. Combined therapy guided by multimodal imaging of fifteen retinal capillary hemangioblastomas in a monocular Von Hippel- Lindau syndrome case report. BMC Ophthalmol. 2022;22(1):205. Figure 4. PMID: 35524216; PMCID: PMC9074324; DOI: 10.1186/s12886-022-02409-8. License: CC BY.
Photographies du fond d’œil montrant un hémangiome capillaire rétinien typique orange-rouge dans la rétine périphérique, avec des vaisseaux nourriciers (artère afférente et veine efférente) nettement dilatés et tortueux, ainsi que l’évolution de la régression tumorale après cryocoagulation. Ces images correspondent aux signes du fond d’œil traités dans la section « 2. Principaux symptômes et signes cliniques ».
L’hémangioblastome rétinien est classé comme suit selon le mode d’apparition et la localisation.
Type périphérique
Fréquence : Forme typique représentant la majorité des cas.
Examen du fond d’œil : Formation d’une tumeur arrondie de couleur rouge orangé dans la périphérie rétinienne, accompagnée d’une artère afférente et d’une veine efférente nettement dilatées et tortueuses.
Au fond d’œil, des tumeurs rétiniennes rondes, solitaires ou multiples, sont caractéristiques. Les lésions périphériques sont associées à des vaisseaux afférents et efférents dilatés et tortueux, et apparaissent généralement avant l’âge de 30 ans. Environ la moitié des cas sont bilatéraux et peuvent survenir à divers endroits du fond d’œil.
La tumeur elle-même est un hémangioblastome composé de capillaires et de cellules stromales spumeuses, produisant une grande quantité de VEGF (facteur de croissance endothélial vasculaire). Le VEGF provoque un décollement de rétine exsudatif, entraînant une baisse de l’acuité visuelle.
Dilatation marquée des vaisseaux conjonctivaux et épiscléraux : dans les cas très avancés, des rapports font état d’une acuité visuelle de NLP et d’une pression intraoculaire de 45 mmHg dans l’œil droit2).
La photographie grand angle du fond d’œil et l’angiographie OCT (OCTA) sont utiles pour l’évaluation des lésions et le suivi 1). L’OCT permet d’évaluer la morphologie de la tumeur, la présence de liquide sous-rétinien et l’œdème maculaire.
La fréquence de la maladie de VHL est d’une personne sur 36 000. Le nombre de patients VHL au Japon est estimé entre 600 et 1 000 cas. La fréquence des hémangiomes rétiniens chez les patients VHL est de 40 à 70 %, et l’âge moyen d’apparition est de 25 ans 1).
Distribution par âge : survient principalement entre 10 et 40 ans. L’apparition avant 10 ans représente environ 5 %.
Bilatéralité : environ la moitié des cas sont bilatéraux.
Multiplicité : plusieurs tumeurs peuvent survenir dans le même œil.
Risque de progression vers VHL dans les cas sporadiques : chez les enfants de moins de 10 ans diagnostiqués comme sporadiques, 45 % reçoivent ultérieurement un diagnostic de maladie de VHL.
Le gène VHL suit un mode de transmission autosomique dominant. Une mutation germinale (premier hit) est présente sur un allèle du gène VHL, et un second hit somatique entraîne la perte de la fonction suppressive tumorale (hypothèse des deux hits).
Comme facteur de risque génétique, tous les membres des familles porteuses d’une mutation pathogène du gène VHL nécessitent une surveillance, et un test génétique VHL doit être envisagé chez les patients atteints d’hémangiome rétinien de moins de 40 ans 1).
L’examen du fond d’œil montre une combinaison caractéristique de tumeurs orange-rouge périphériques et de vaisseaux afférents et efférents dilatés et tortueux. L’évaluation combine les examens suivants :
Photographie du fond d’œil grand angle : utile pour visualiser l’ensemble des lésions périphériques.
Angiographie à la fluorescéine (FA) : identification de la tumeur et évaluation de l’activité. Une fuite fluorescente précoce et abondante est un critère diagnostique.
OCT/OCTA : évaluation morphologique de la tumeur, quantification de l’œdème maculaire et du liquide sous-rétinien.
Échographie (mode B) : confirmation d’une tumeur solide et évaluation du décollement de la rétine.
Dans les familles VHL, l’examen du fond d’œil commence dès la naissance (0 an) et se poursuit au moins une fois par an 1). La combinaison d’une caméra non mydriatique et d’une photographie grand angle permet d’éviter de manquer des lésions périphériques.
Dans la maladie de VHL, des lésions multiples peuvent survenir dans d’autres organes que la rétine, d’où la nécessité d’un bilan systémique régulier par une équipe pluridisciplinaire 1).
IRM cérébrale : dépistage des hémangioblastomes du cervelet, du tronc cérébral et de la moelle épinière.
Échographie abdominale, TDM/IRM : recherche de carcinome rénal, de kystes pancréatiques, rénaux et surrénaliens.
Dépistage du phéochromocytome : dosage des catécholamines urinaires et de leurs métabolites.
La recherche systémique ci-dessus doit être poursuivie environ une fois par an.
Le test du gène VHL est indiqué chez les patients atteints de tumeur vasculaire rétinienne sporadique âgés de moins de 40 ans, ainsi que chez les patients et leurs familles suspectés de maladie de VHL 1). Il est recommandé de le réaliser en parallèle avec un conseil génétique.
Un diagnostic différentiel avec les maladies suivantes est nécessaire.
Maladie de Coats : fréquente chez les garçons. Pas de dilatation ni de tortuosité des vaisseaux afférents et efférents, mais présence d’exsudats durs et de décollement de rétine exsudatif.
Syndrome de Wyburn-Mason (angiome racémeux) : association de malformations artérioveineuses rétiniennes et cérébrales.
Hémangiome choroïdien : lésion orange-rouge, mais sans complications systémiques de la maladie de VHL.
Tumeur vasoproliférative rétinienne : prédominance dans la périphérie inférieure. À distinguer des modifications secondaires.
QOù trouve-t-on les signes du fond d'œil de l'hémangiome rétinien ?
A
Le type périphérique survient souvent dans la périphérie rétinienne, nécessitant un examen détaillé du fond d’œil sous dilatation. Les petites tumeurs précoces peuvent ressembler à des microanévrismes ; l’utilisation de la photographie grand angle du fond d’œil peut éviter de les négliger. Le type papillaire apparaît comme une masse péripapillaire, et l’identification des vaisseaux afférents et efférents est souvent difficile.
Le traitement est choisi en fonction de la localisation, de la taille et du degré de modifications exsudatives de l’hémangiome. Comme l’hémangiome devient difficile à traiter lorsqu’il augmente de taille, un diagnostic et un traitement précoces sont importants. Un traitement précoce des lésions de petite taille peut améliorer le pronostic visuel.
Indications : Traitement de première intention pour le type périphérique. Pour les lésions de moins d’un diamètre papillaire, une guérison complète peut être attendue.
Méthode : La coagulation directe de l’hémangiome est répétée jusqu’à obtention d’un effet suffisant. Une photocoagulation précoce est également recommandée pour les petites lésions de moins d’un diamètre papillaire1).
Limites : Pour les lésions volumineuses dépassant un diamètre papillaire, plusieurs séances de traitement sont nécessaires.
Cryocoagulation
Indications : Lésions volumineuses ou cas où la photocoagulation au laser est difficile d’accès.
Méthode : La cryocoagulation est choisie en fonction de la taille et du degré de saillie de l’hémangiome.
En cas de modifications exsudatives associées : Drainage du liquide sous-rétinien, diathermie, etc., sont combinés, mais le traitement est difficile.
PDT et thérapie anti-VEGF : Récemment, des rapports de cas se sont accumulés sur la thérapie photodynamique (PDT) et les agents anti-VEGF (bévacizumab, ranibizumab, etc.) en monothérapie ou en association3). Ils sont utilisés pour supprimer les modifications exsudatives de manière adjuvante, mais ne sont pas couverts par l’assurance maladie et nécessitent une décision au cas par cas.
Le traitement du type papillaire n’est pas encore établi1). La photocoagulation au laser comporte un risque élevé de lésions du nerf optique et de la macula, et l’indication doit être évaluée avec une prudence particulière. Les traitements suivants ont été rapportés dans des études de cas, mais aucun n’est établi comme traitement standard.
Injections intravitréennes d’agents anti-VEGF : Un effet suppressif sur les modifications exsudatives est attendu, mais l’effet de réduction tumorale est limité.
Radiothérapie par protons : Irradiation concentrée sur la tumeur5).
Une évaluation minutieuse du moment de l’intervention est nécessaire, et une conférence multidisciplinaire dans un établissement spécialisé est recommandée.
Après le traitement, les récidives et les nouvelles lésions sont fréquentes, donc une surveillance à vie est nécessaire. Après chaque traitement, un examen du fond d’œil doit être effectué dans les 3 à 6 mois pour vérifier l’efficacité et détecter d’éventuelles nouvelles lésions. Dans la maladie de VHL, plusieurs lésions apparaissent à des moments différents, il ne faut donc pas négliger l’examen de toute la périphérie rétinienne.
Pour le pronostic vital de la maladie de VHL, la gestion du carcinome rénal et de l’hémangioblastome du système nerveux central est importante. En collaboration multidisciplinaire avec la neurochirurgie, l’urologie, l’endocrinologie, etc., une surveillance et des interventions appropriées doivent être effectuées pour chaque lésion organique.
6. Physiopathologie et mécanismes détaillés de la pathogenèse
Le gène VHL est un gène suppresseur de tumeur situé sur le chromosome 3p25-26, codant pour la protéine pVHL, un composant du complexe E3 ubiquitine ligase. La fonction principale de la protéine pVHL est l’ubiquitination et la dégradation protéasomique de la sous-unité alpha du facteur induit par l’hypoxie (HIF).
Dans des conditions normales, HIFα est reconnu par pVHL, ubiquitiné et rapidement dégradé. Lorsque le gène VHL est inactivé, la fonction de pVHL est perdue et HIFα s’accumule.
Formation tumorale selon l’hypothèse du double hit
Dans la maladie de VHL, en plus de la mutation germinale (1er hit), une perte complète de la fonction du gène VHL due à un second hit somatique (perte d’hétérozygotie, etc.) conduit à la formation de tumeurs.
Lorsque HIFα s’accumule dans le noyau, la transcription de nombreux gènes liés à l’angiogenèse et à la prolifération cellulaire, tels que VEGF, PDGF et EPO, est activée de manière constitutive. Dans la maladie de VHL, HIF-2α (EPAS1) fonctionne comme le principal moteur 1).
Le rétinoblastome vasculaire est composé de deux types de cellules.
Cellules stromales spumeuses : riches en lipides, elles produisent de nombreuses cytokines, dont le VEGF.
Capillaires : induits secondairement par le VEGF produit par les cellules stromales.
Ces cellules stromales constituent la tumeur elle-même, et la production constante de VEGF est la cause directe de la prolifération vasculaire tumorale et du décollement de rétine exsudatif.
Les associations entre les types de mutations du gène VHL et les phénotypes cliniques (type 1 : non associé au phéochromocytome, type 2 : associé au phéochromocytome, etc.) sont partiellement connues, mais une analyse plus détaillée de la classification et des différences de fréquence des lésions oculaires est attendue.
Le belzutifan est un composé de faible poids moléculaire qui inhibe sélectivement HIF-2α. En 2021, la FDA (Food and Drug Administration américaine) l’a approuvé pour les patients atteints de la maladie de VHL présentant un carcinome rénal métastatique, un hémangioblastome du système nerveux central ou un rétinoblastome vasculaire 7).
Les résultats de l’essai de phase 2 (LITESPARK-004) concernant les hémangiomes rétiniens sont les suivants 1).
Taux de réponse : 100 % (12/12 cas) ont montré une réduction tumorale.
Carcinome rénal : réponse chez 49 % des patients.
Hémangioblastome du système nerveux central : réponse chez 30 % des patients.
Les principaux effets indésirables rapportés sont l’anémie (environ 90 %) et la fatigue (environ 66 %). Des essais cliniques au Japon sont également prévus, et ce traitement est considéré comme un candidat prometteur pour le futur traitement standard 1).
Dans l’essai de phase 2 rapporté par Jonasch et al. (2021), l’administration orale de belzutifan 120 mg une fois par jour chez des patients atteints de la maladie de VHL a montré un effet de réduction de 100 % sur les hémangioblastomes rétiniens 7).
La thérapie photodynamique (PDT) a été rapportée comme étant applicable aussi bien aux lésions périphériques qu’à celles de la tête du nerf optique.
di Nicola et al. (2022) ont rapporté l’efficacité de la PDT pour les hémangioblastomes rétiniens, montrant notamment son applicabilité aux lésions juxtapapillaires 4).
Schmidt-Erfurth et al. ont évalué l’applicabilité de la PDT aux lésions de la tête du nerf optique et les risques de complications 6).
Hussain et al. ont rapporté l’effet de la radiothérapie par protons pour les hémangiomes capillaires rétiniens juxtapapillaires 5). Des séries de cas s’accumulent en tant qu’option pour les lésions difficiles à traiter de la tête du nerf optique.
L’effet de réduction directe de la tumeur est limité, mais les rapports de cas utilisant ce traitement comme contrôle adjuvant des modifications exsudatives augmentent 1). Le protocole n’est pas établi et une décision au cas par cas est nécessaire.
QLe belzutifan est-il disponible au Japon ?
A
En avril 2026, le belzutifan n’est pas approuvé pour une utilisation générale au Japon. La FDA l’a approuvé en 2021, mais au Japon, il est en phase de préparation d’essais cliniques 1). Pour les patients diagnostiqués avec la maladie de VHL pour lesquels le traitement standard est difficile, envisager de consulter le médecin traitant ou un centre spécialisé pour une éventuelle participation à un essai clinique.
QQue se passe-t-il si un hémangioblastome rétinien n'est pas traité ?
A
Si l’hémangiome augmente de taille, il devient réfractaire et le pronostic visuel se détériore considérablement. Si le décollement de rétine exsudatif atteint la macula, la perte de vision peut devenir irréversible, et une progression vers un décollement de rétine tractionnel ou un glaucome néovasculaire peut conduire à la cécité. Dans la maladie de VHL, de nouvelles lésions apparaissent tout au long de la vie, donc un examen du fond d’œil continu est indispensable en parallèle du traitement.
Lin H, Lin X. Pronounced conjunctival vascular engorgement in von Hippel-Lindau syndrome: a case report. BMC Ophthalmol. 2020 (症例報告).
Krivosic V, Kamami-Levy C, Jacob J, Richard S, Tadayoni R, Gaudric A.. Laser Photocoagulation for Peripheral Retinal Capillary Hemangioblastoma in von Hippel-Lindau Disease. Ophthalmol Retina. 2017;1(1):59-67. doi:10.1016/j.oret.2016.08.004. PMID:31047395.
Di Nicola M, Williams BK Jr, Hua J, Bekerman VP, Mashayekhi A, Shields JA, Shields CL. Photodynamic Therapy for Retinal Hemangioblastoma: Treatment Outcomes of 17 Consecutive Patients. Ophthalmology. Retina. 2022;6(1):80-88. doi:10.1016/j.oret.2021.04.007. PMID:33892136.
Hussain RN, Jmor F, Damato B, et al. Proton beam radiotherapy for retinal capillary haemangioblastoma. Br J Ophthalmol. 2016;100(3):317-321.
Schmidt-Erfurth UM, Kusserow C, Barbazetto IA, Laqua H.. Benefits and complications of photodynamic therapy of papillary capillary hemangiomas. Ophthalmology. 2002;109(7):1256-1266. doi:10.1016/s0161-6420(02)01059-x. PMID:12093647.
Jonasch E, Donskov F, Iliopoulos O, Rathmell WK, Narayan VK, Maughan BL, Oudard S, Else T, Maranchie JK, Welsh SJ, Thamake S, Park EK, Perini RF, Linehan WM, Srinivasan R, MK-6482-004 Investigators.. Belzutifan for Renal Cell Carcinoma in von Hippel-Lindau Disease. N Engl J Med. 2021;385(22):2036-2046. doi:10.1056/nejmoa2103425. PMID:34818478; PMCID:PMC9275515.
Copiez le texte de l'article et collez-le dans l'assistant IA de votre choix.
Article copié dans le presse-papiers
Ouvrez un assistant IA ci-dessous et collez le texte copié dans la conversation.