L’hamartome astrocytaire rétinien est une tumeur bénigne résultant de la prolifération excessive des astrocytes de la rétine. Un hamartome est une lésion tumorale composée de tissus matures normalement présents dans la région, mais dans des proportions anormales. Il n’y a pas de risque de transformation maligne.
Cette affection peut être associée à la sclérose tubéreuse de Bourneville (TSC) ou survenir de façon sporadique sans TSC. La sclérose tubéreuse est une maladie multisystémique autosomique dominante caractérisée par des hamartomes dans divers organes, se manifestant par des crises d’épilepsie dues à des lésions intracrâniennes, des adénomes sébacés cutanés, des angiomyolipomes rénaux et des hamartomes rétiniens.
Deux gènes sont impliqués : TSC1 (chromosome 9) et TSC2 (chromosome 16). TSC1 code pour l’hamartine et TSC2 pour la tubérine, et leur dysfonctionnement entraîne une dérégulation de la voie mTOR (mechanistic target of rapamycin).
QL'hamartome astrocytaire rétinien peut-il survenir en l'absence de sclérose tubéreuse ?
A
Oui, des cas sporadiques sans TSC existent. Dans ces cas, l’atteinte bilatérale est rare et, en l’absence de complications systémiques, la prise en charge est principalement ophtalmologique. En revanche, en cas d’association à la TSC, une gestion multidisciplinaire est nécessaire, d’où l’importance de différencier la TSC lors du diagnostic.
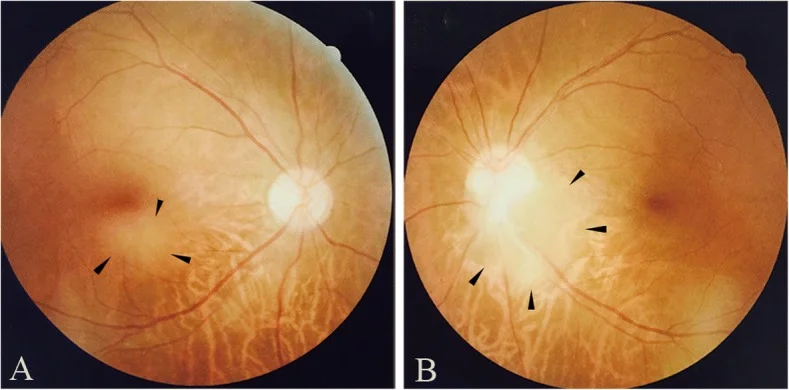
Qin X, Tao Y, Zhang Z. Retinal astrocytic hamartoma in tuberous sclerosis complex in an elderly person: a case report. BMC Ophthalmol. 2018;18(1):319. Figure 1. PMID: 30541513; PMCID: PMC6292060; DOI: 10.1186/s12886-018-0991-z. License: CC BY.
Photographies du fond d’œil montrant un astrocytome rétinien dans la région maculaire inférieure de l’œil droit (a) et au bord temporal inférieur de la papille optique de l’œil gauche (b). Correspond aux signes du fond d’œil traités dans la section « 2. Principaux symptômes et signes cliniques ».
L’astrocytome rétinien est souvent asymptomatique et découvert fortuitement lors d’un examen de dépistage du fond d’œil pour la sclérose tubéreuse. Rarement, une baisse de l’acuité visuelle ou des corps flottants peuvent survenir. Lorsque la tumeur est située dans la région maculaire, l’impact sur la vision est important.
Le fond d’œil présente des lésions blanchâtres surélevées caractéristiques. Selon leur apparence, elles sont classées en deux types suivants.
Type mûrier (mulberry)
Aspect : Élévation en dôme avec une surface irrégulière. Forme caractéristique évoquant une « mûre ».
Calcification : Souvent associée à une calcification. Par rapport à la calcification du rétinoblastome, elle se caractérise par une teinte jaunâtre plus prononcée.
Bords : Reflet d’une division cellulaire lente, l’élévation est légère et les bords sont en pente douce.
Type plat
Aspect : Lésion blanchâtre translucide et plate, d’apparence similaire à un rétinoblastome.
Importance du diagnostic différentiel : En raison de la ressemblance avec le rétinoblastome, une évaluation minutieuse est nécessaire pour le diagnostic différentiel.
Calcification : Souvent sans calcification par rapport au type mûrier.
L’angiographie à la fluorescéine (FAG) montre une visualisation précoce des microvaisseaux intratumoraux. L’absence de fuite tardive de fluorescéine est une caractéristique de cette maladie et constitue un point important de différenciation avec le rétinoblastome, qui présente une fuite tardive.
En tomographie par cohérence optique (OCT), la tumeur apparaît comme une masse hyperréflective surélevée depuis les couches internes de la rétine. Les caractéristiques incluent une désorganisation des couches internes de la tumeur et une limite relativement nette avec la rétine normale environnante.
La sclérose tubéreuse de Bourneville (STB) suit un mode de transmission autosomique dominant. Sa prévalence est d’environ 1 personne sur 6 000 à 10 000. Le taux de comorbidité des hamartomes rétiniens chez les patients atteints de STB est d’environ 50 %, et ils peuvent être bilatéraux et multiples.
Les mutations du gène TSC1 (chromosome 9q34) sont associées à des symptômes plus légers, tandis que les mutations du gène TSC2 (chromosome 16p13.3) tendent à provoquer des lésions systémiques plus graves. Les mutations de novo sont fréquentes, donc l’absence d’antécédents familiaux n’exclut pas la STB.
Dans les cas sporadiques (non STB), le contexte génétique est différent et des mutations somatiques localisées peuvent être impliquées. Les cas sporadiques sont généralement unilatéraux et uniques, sans complications systémiques.
Antécédents familiaux ou diagnostic confirmé de sclérose tubéreuse de Bourneville (STB)
Mutations génétiques connues de la STB (TSC1 ou TSC2)
Antécédents de lésions multiples du système nerveux central, cutanées ou rénales
QExiste-t-il une prédisposition liée au sexe ou à l'âge ?
A
Aucune différence significative entre les sexes n’a été rapportée. Dans les cas associés à la STB, les lésions du fond d’œil peuvent être détectées dès la petite enfance, et un examen du fond d’œil est recommandé dans le cadre du dépistage pédiatrique. Les cas sporadiques peuvent être découverts chez l’adulte, mais tous suivent une évolution bénigne.
Le diagnostic de l’astrocytome rétinien repose sur la combinaison des signes caractéristiques du fond d’œil et des manifestations systémiques de la sclérose tubéreuse.
Points clés de l’examen du fond d’œil pour le diagnostic différentiel :
Lésion blanchâtre surélevée d’aspect mûriforme ou plat
Absence de fuite tardive de fluorescéine à l’angiographie
Surélévation légère avec bords réguliers
Absence de croissance lors du suivi
Teinte jaunâtre même en présence de calcifications
La principale maladie à différencier est le rétinoblastome. Chez l’enfant présentant une lésion rétinienne blanchâtre surélevée, il faut d’abord exclure un rétinoblastome.
Lorsqu’un hamartome rétinien est découvert, une évaluation systémique doit être réalisée selon les critères diagnostiques de la TSC (révision de Northrup 2012 1)). Les critères majeurs incluent l’adénome sébacé (fibrome angio-facial), l’épilepsie, les nodules cérébraux (taches de Shagreen), etc. Une collaboration avec les services de pédiatrie et de neurologie est essentielle pour un diagnostic définitif de TSC.
L’hamartome rétinien à cellules astrocytaires ne grossit généralement pas ; en l’absence de symptômes ou d’augmentation de taille, aucun traitement n’est nécessaire et la surveillance est la règle. Un examen du fond d’œil régulier est effectué pour vérifier l’absence de croissance ou d’hémorragie.
Indications thérapeutiques et techniques chirurgicales
Si la lésion oculaire n’augmente pas, un traitement actif n’est pas nécessaire. Chez les patients atteints de sclérose tubéreuse, la gestion des lésions systémiques (épilepsie, astrocytome sous-épendymaire à cellules géantes SEGA, angiomyolipome rénal) doit être effectuée en collaboration avec un pédiatre et un neurologue.
Inhibiteurs de mTOR (évérolimus) sont approuvés pour les tumeurs systémiques associées à la sclérose tubéreuse (SEGA et angiomyolipome rénal) et ont montré un effet de réduction tumorale dans le cadre du traitement systémique de la TSC2)3). Les preuves d’efficacité directe sur les hamartomes rétiniens sont limitées, mais des cas de réduction ont été rapportés chez des patients traités pour la TSC systémique.
QUn traitement de l'hamartome rétinien à cellules astrocytaires est-il nécessaire ?
A
En l’absence de symptômes et de progression, aucun traitement n’est nécessaire ; une surveillance régulière par examen du fond d’œil est la règle. Une vitrectomie ou une photocoagulation rétinienne n’est réalisée qu’en cas d’hémorragies récurrentes. Chez les patients atteints de sclérose tubéreuse, la prise en charge des maladies systémiques est importante en parallèle du suivi ophtalmologique.
La pathogénie de l’hamartome rétinien à cellules astrocytaires résulte d’une perte de fonction des produits des gènes TSC1 (hamartine) et TSC2 (tubérine).
L’hamartine et la tubérine forment un complexe qui agit comme suppresseur de tumeur en inhibant l’activité du complexe mTORC1 (mechanistic target of rapamycin complex 1). Les mutations de TSC1 ou TSC2 entraînent une perte de ce contrôle, conduisant à une hyperactivation de mTORC1.
L’hyperactivation de mTORC1 favorise la prolifération cellulaire, la synthèse protéique et l’angiogenèse via la phosphorylation de S6 kinase (S6K) et de 4E-BP1. Cela entraîne une prolifération anormale des astrocytes et la formation d’hamartomes (tumeurs bénignes).
On pense que l’hypothèse des deux coups (2-hit hypothesis) est impliquée dans le développement tumoral. Chez les individus porteurs d’une mutation allélique de TSC1 ou TSC2 dans la lignée germinale, une mutation somatique (second hit) dans les cellules rétiniennes entraîne la perte de fonction de l’allèle normal restant (LOH : perte d’hétérozygotie), ce qui conduit à un dérèglement complet de la voie mTOR et à la formation de tumeurs.
L’hamartome astrocytaire rétinien est une tumeur bénigne constituée d’une prolifération de cellules astrocytaires, sans transformation maligne. Histologiquement, les astrocytes anormalement proliférés sont densément arrangés, avec parfois des dépôts de calcium (calcification). La faible vitesse de division cellulaire correspond à la caractéristique clinique d’une croissance tumorale lente.
L’évérolimus (everolimus), un inhibiteur de mTORC1, est utilisé dans le traitement des tumeurs associées à la sclérose tubéreuse. Dans l’essai EXIST-1 (ciblant les SEGA) 2), la proportion de patients ayant présenté une réduction d’au moins 50 % du volume des SEGA était significativement plus élevée dans le groupe évérolimus que dans le groupe placebo. L’essai EXIST-2 (ciblant les angiomyolipomes rénaux) 3) a également rapporté un effet similaire de réduction tumorale.
En ce qui concerne l’efficacité ophtalmique des inhibiteurs de mTOR sur les hamartomes rétiniens associés à la TSC, seuls quelques rapports de cas limités existent à ce jour. Des réductions d’hamartomes rétiniens ont été observées chez des patients traités par évérolimus dans le cadre d’un traitement systémique de la TSC, mais aucune preuve issue d’essais prospectifs à grande échelle n’a été établie.
Les hamartomes du fond d’œil ne grossissent généralement pas, mais dans de rares cas, ils peuvent augmenter de volume et provoquer des hémorragies ou des exsudats. L’identification des facteurs de risque de croissance, la prédiction des changements de vision à long terme et la détermination du moment approprié pour une intervention nécessitent une accumulation supplémentaire de cas et des études prospectives.
Pathologie de l’hamartome astrocytaire rétinien sporadique
Le contexte génétique et les mécanismes moléculaires des cas sporadiques sans TSC ne sont pas encore entièrement élucidés. Il est suggéré qu’une anomalie locale de la voie mTOR due à des mutations somatiques pourrait être impliquée, mais l’élucidation des mécanismes détaillés est une question de recherche future.
Rowley SA, O’Callaghan FJ, Osborne JP.. Ophthalmic manifestations of tuberous sclerosis: a population based study. Br J Ophthalmol. 2001;85(4):420-423. doi:10.1136/bjo.85.4.420. PMID:11264130; PMCID:PMC1723924.
Zimmer-Galler IE, Robertson DM.. Long-term observation of retinal lesions in tuberous sclerosis. Am J Ophthalmol. 1995;119(3):318-324. doi:10.1016/s0002-9394(14)71174-2. PMID:7872393.
Neil J Lucchese, Morton F Goldberg. Iris and Fundus Pigmentary Changes in Tuberous Sclerosis. J Pediatr Ophthalmol Strabismus. 1981;18(6):45-46. doi:10.3928/0191-3913-19811101-12.
Copiez le texte de l'article et collez-le dans l'assistant IA de votre choix.
Article copié dans le presse-papiers
Ouvrez un assistant IA ci-dessous et collez le texte copié dans la conversation.