
Hémangiome choroïdien diffus
1. Qu’est-ce que l’hémangiome choroïdien diffus ?
Section intitulée « 1. Qu’est-ce que l’hémangiome choroïdien diffus ? »Définition et concept
Section intitulée « Définition et concept »Il existe deux types d’hémangiome choroïdien : localisé (solitaire) et diffus. L’hémangiome localisé est bien délimité et survient de manière sporadique, tandis que l’hémangiome diffus est souvent étendu et mal délimité. L’hémangiome choroïdien diffus est presque toujours associé au syndrome de Sturge-Weber (angiomatose encéphalo-trigéminée).
Le syndrome de Sturge-Weber est une phacomatose (syndrome neurocutané) causée par une prolifération ectopique des cellules de la crête neurale au cours de la vie embryonnaire. Il se caractérise par la formation d’hémangiomes au niveau du visage, de l’œil et de la pie-mère cérébrale.
Épidémiologie
Section intitulée « Épidémiologie »La fréquence du syndrome de Sturge-Weber est estimée à 1 personne sur 20 000 à 50 000. Il n’est pas héréditaire ; la cause principale est une mutation somatique mosaïque du gène GNAQ survenant de manière sporadique. Il s’agit d’une maladie congénitale présente dès la naissance, détectée dès la petite enfance. Elle survient dans l’œil ipsilatéral à un angiome cutané (tache de vin) présent dans le territoire des première et deuxième branches du nerf trijumeau facial. Les données sur les différences de sexe et de race sont limitées, mais en tant que maladie sporadique, elle peut survenir dans toutes les races.
Comparaison avec l’hémangiome choroïdien localisé
Section intitulée « Comparaison avec l’hémangiome choroïdien localisé »| Caractéristique | Hémangiome choroïdien diffus | Hémangiome choroïdien localisé (solitaire) |
|---|---|---|
| Syndrome associé | Toujours associé au syndrome de Sturge-Weber | Aucune maladie systémique (sporadique) |
| Distribution au fond d’œil | Étendue, limites floues | Localisée, limites relativement nettes |
| Couleur | Fond d’œil en ketchup de tomate | Lésion surélevée orange |
| Association au glaucome | Plus de la moitié | Rare |
| Âge d’apparition | Congénital / petite enfance | Souvent à l’âge adulte |
L’hémangiome choroïdien diffus survient presque toujours en association avec le syndrome de Sturge-Weber (angiomatose encéphalotrigéminée). Le syndrome de Sturge-Weber se caractérise par une triade : tache de vin de Porto faciale, angiome leptoméningé et symptômes ophtalmiques (hémangiome choroïdien diffus, glaucome). Sa fréquence est de 1 pour 20 000 à 50 000 personnes et il n’est pas héréditaire. L’hémangiome choroïdien apparaît du même côté que la tache de vin de Porto faciale.
2. Principaux symptômes et signes cliniques
Section intitulée « 2. Principaux symptômes et signes cliniques »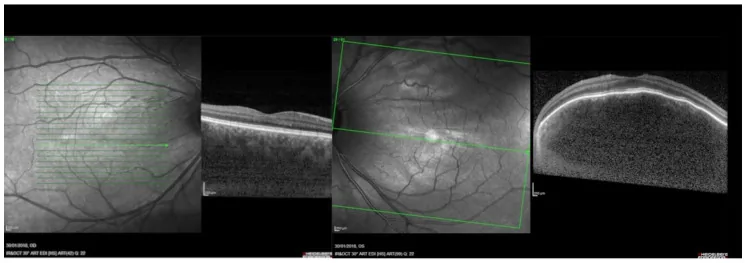
Signes du fond d’œil
Section intitulée « Signes du fond d’œil »Le signe le plus caractéristique de l’hémangiome choroïdien diffus est un fond d’œil de couleur rouge orangé sur une large zone, appelé « fond d’œil en ketchup ». La tumeur occupe une grande partie de la choroïde et ses limites sont floues. Sur une photographie standard du fond d’œil, la différence de rougeur est nettement visible par rapport à un fond d’œil normal.
Les principales observations du fond d’œil et ophtalmologiques sont les suivantes :
- Épaississement choroïdien diffus : la choroïde est épaissie de manière étendue du pôle postérieur à la périphérie
- Limites floues : contrairement à l’hémangiome circonscrit, les bords de la tumeur sont mal définis
- Décollement séreux de la rétine : principale cause de baisse de l’acuité visuelle. Se manifeste par des troubles du champ visuel et une métamorphopsie
- Hypermétropisation : le soulèvement choroïdien entraîne un changement de réfraction dans l’axe oculaire
- Lésion surélevée orange : reconnue comme une surélévation orange au niveau du pôle postérieur
Angiographie fluorescéinique et imagerie
Section intitulée « Angiographie fluorescéinique et imagerie »Les signes caractéristiques des différents examens d’imagerie sont présentés ci-dessous.
- FA (angiographie à la fluorescéine) : hyperfluorescence réticulaire précoce dans la tumeur, avec augmentation de la rétention du colorant en phase tardive (motif diffus)
- ICGA (angiographie au vert d’indocyanine) : meilleure visualisation des vaisseaux choroïdiens qu’à la FA, permettant une identification claire de la structure vasculaire tumorale
- OCT : observé comme un soulèvement localisé étendu de la choroïde postérieure. Utile également pour évaluer le décollement séreux de la rétine
- Échographie : visualisé comme une lésion surélevée solide. Le B-scan confirme un épaississement choroïdien étendu
- CT : visualisé comme une lésion solide, similaire à l’échographie
Association avec le glaucome
Section intitulée « Association avec le glaucome »Dans le syndrome de Sturge-Weber, plus de la moitié des cas présentent un glaucome. Selon l’âge d’apparition, on observe environ la moitié des cas de glaucome infantile (type congénital) et l’autre moitié de glaucome juvénile (type acquis).
- Type d’apparition infantile : présente les caractéristiques du glaucome congénital telles que l’élargissement du diamètre cornéen, le buphtalmie (élargissement cornéen) et les stries de Haab. Une intervention chirurgicale est souvent nécessaire.
- Type d’apparition juvénile : la pression intraoculaire se stabilise d’abord dans la plage normale, puis une augmentation de la pression peut survenir lors du suivi.
- Hémangiome épiscléral : certains cas présentent un hémangiome de l’épisclère du côté affecté.
Plus de la moitié des cas de syndrome de Sturge-Weber présentent un glaucome associé. L’apparition infantile et juvénile représentent chacune la moitié des cas, et une augmentation tardive de la pression intraoculaire peut survenir lors du suivi. Sans une gestion appropriée, le glaucome peut entraîner des troubles du champ visuel et une baisse de l’acuité visuelle, d’où la nécessité d’une mesure régulière de la pression intraoculaire et d’un examen du fond d’œil à long terme. En particulier, la buphtalmie et l’opacité cornéenne chez le nourrisson nécessitent une intervention urgente.
3. Causes et facteurs de risque
Section intitulée « 3. Causes et facteurs de risque »Mécanisme pathogénique
Section intitulée « Mécanisme pathogénique »Le syndrome de Sturge-Weber et l’hémangiome choroïdien diffus sont causés par une prolifération ectopique des cellules de la crête neurale pendant la période embryonnaire. Un hamartome vasculaire choroïdien se forme, remplaçant largement la structure choroïdienne normale par un tissu vasculaire.
Au niveau génétique, une mutation du gène GNAQ (protéine Gqα liant les nucléotides guanine) (c.548G>A, p.Arg183Gln) est détectée dans de nombreux cas. Cette mutation survient sous forme de mutation somatique en mosaïque, elle n’est donc pas héréditaire et n’est généralement pas transmise des parents aux enfants. On pense que la mutation GNAQ réduit l’activité GTPase et active de manière constitutive les signaux de formation vasculaire.
Principaux facteurs de risque et caractéristiques
Section intitulée « Principaux facteurs de risque et caractéristiques »- Mutation somatique en mosaïque de GNAQ : la plus importante comme mutation causale. Il ne s’agit pas d’une mutation germinale, donc non héréditaire.
- Maladie congénitale : présente dès la naissance, principalement due à une mutation accidentelle pendant la période embryonnaire plutôt qu’à un contexte génétique.
- Facteurs environnementaux : aucun facteur de risque environnemental établi.
- Antécédents familiaux : en principe non héréditaire, donc aucune accumulation familiale habituellement observée.
Mécanisme de développement du glaucome
Section intitulée « Mécanisme de développement du glaucome »Deux mécanismes principaux sont impliqués dans le développement du glaucome dans le syndrome de Sturge-Weber.
- Augmentation de la pression veineuse épisclérale : L’angiome épiscléral augmente la pression veineuse, ce qui accroît la résistance à l’écoulement de l’humeur aqueuse. Mécanisme fréquent dans le glaucome survenant après l’enfance.
- Anomalie de l’angle : Une anomalie du développement de l’angle de la chambre antérieure entrave l’écoulement de l’humeur aqueuse. Souvent impliqué dans le glaucome à début infantile.
4. Diagnostic et méthodes d’examen
Section intitulée « 4. Diagnostic et méthodes d’examen »Approche diagnostique
Section intitulée « Approche diagnostique »L’association d’une tache de vin (angiome cutané dans le territoire du nerf trijumeau) et de modifications du fond d’œil du même côté soutient fortement le diagnostic d’angiome choroïdien diffus. Les examens suivants sont utilisés pour le diagnostic ophtalmologique.
Examen du fond d’œil et imagerie :
- Photographie du fond d’œil (y compris l’angiographie grand angle) : enregistrement et suivi de l’aspect « fond d’œil ketchup ».
- Angiographie à la fluorescéine (FA) / angiographie au vert d’indocyanine (ICGA) : confirmation d’une hyperfluorescence réticulaire précoce (motif diffus).
- Tomographie par cohérence optique (OCT) : évaluation d’un épaississement choroïdien étendu et d’un décollement séreux de la rétine.
- Échographie (mode A et B) : confirmation d’une lésion solide et mesure de son épaisseur.
Bilan systémique :
- IRM cérébrale : leptoméningiome angiomateux et calcifications (évalués par un neuropédiatre en parallèle des examens ophtalmologiques)
- Tonométrie : dépistage du glaucome (sous anesthésie générale si nécessaire)
- Gonioscopie : évaluation des anomalies de l’angle iridocornéen
Diagnostic différentiel
Section intitulée « Diagnostic différentiel »Les maladies nécessitant un diagnostic différentiel avec l’hémangiome choroïdien diffus sont présentées ci-dessous.
| Maladie différentielle | Principales différences |
|---|---|
| Hémangiome choroïdien circonscrit | Limites nettes, localisé, pas de syndrome systémique |
| Mélanome malin de la choroïde (forme diffuse) | Signes malins, modifications pigmentaires, risque métastatique |
| Uvéite postérieure | Signes inflammatoires, opacités vitréennes |
| Variation de couleur du fond d’œil normal | Absence de signes systémiques du syndrome de Sturge-Weber |
Points à considérer lors du diagnostic
Section intitulée « Points à considérer lors du diagnostic »Si le fond d’œil semble « anormalement rouge », en particulier chez les nourrissons présentant une tache lie-de-vin, il faut fortement suspecter cette maladie. Chez les nourrissons, un examen sous anesthésie générale peut être nécessaire, et une collaboration avec les services d’anesthésiologie et de pédiatrie est importante.
5. Traitement standard
Section intitulée « 5. Traitement standard »Aperçu de la stratégie thérapeutique
Section intitulée « Aperçu de la stratégie thérapeutique »Le traitement de l’hémangiome choroïdien diffus est effectué par étapes en fonction de la présence ou non de symptômes et du type de complications. L’ablation chirurgicale de l’hémangiome choroïdien lui-même n’est généralement pas pratiquée. Les principales cibles du traitement sont le décollement séreux de la rétine (principale cause de baisse de vision) et le glaucome.
1. Surveillance
Section intitulée « 1. Surveillance »En l’absence de symptômes et si la vision et la pression intraoculaire sont normales, la surveillance régulière suivante est poursuivie :
- Mesure de la pression intraoculaire (détection précoce du glaucome)
- Photographie du fond d’œil et OCT pour vérifier la présence d’un décollement séreux de la rétine
- Examen de l’acuité visuelle et de la réfraction (évaluation de l’hypermétropie et de l’amblyopie)
- Surveillance par angiographie à la fluorescéine (FA) ou OCT si nécessaire
2. Traitement du décollement séreux de la rétine et de la baisse de la fonction visuelle
Section intitulée « 2. Traitement du décollement séreux de la rétine et de la baisse de la fonction visuelle »En cas de baisse de l’acuité visuelle ou de décollement séreux de la rétine, un traitement actif est effectué.
PDT (thérapie photodynamique) :
- Administration intraveineuse de vertéporfine (photosensibilisant) suivie d’une irradiation laser à 689 nm
- L’efficacité de la PDT pour l’hémangiome choroïdien a été rapportée
- Non couvert par l’assurance maladie (pour l’hémangiome choroïdien diffus)
- On peut s’attendre à une résolution du décollement séreux de la rétine et à une amélioration de l’acuité visuelle
- L’efficacité pour l’hémangiome choroïdien circonscrit est établie, mais l’application pour la forme diffuse présente des limitations techniques en raison de la large zone d’irradiation
Radiothérapie à faible dose :
- Une irradiation à faible dose d’environ 20 Gy est administrée
- Peut être efficace. On peut s’attendre à une résolution du décollement de la rétine et à une amélioration de l’acuité visuelle
- La téléréthérapie (irradiation externe) est souvent utilisée
L’ablation chirurgicale n’est généralement pas pratiquée. L’hémangiome choroïdien diffus s’étend largement sur toute la choroïde, ce qui rend l’ablation chirurgicale difficile tant sur le plan technique que fonctionnel. Pour le décollement séreux de la rétine entraînant une baisse de l’acuité visuelle, la PDT ou la radiothérapie à faible dose (environ 20 Gy) sont réalisées et sont souvent efficaces. Pour le glaucome, la pression intraoculaire est contrôlée par des collyres ou une intervention chirurgicale.
3. Traitement du glaucome
Section intitulée « 3. Traitement du glaucome »Étant donné que plus de la moitié des cas présentent un glaucome associé, sa prise en charge est un enjeu thérapeutique important.
Traitement médicamenteux (collyres) :
- Analogues des prostaglandines (inhibition de la production d’humeur aqueuse, facilitation de l’écoulement)
- Bêta-bloquants (inhibition de la production d’humeur aqueuse)
- Inhibiteurs de l’anhydrase carbonique (topiques ou oraux)
- Cependant, dans le glaucome dû à une pression veineuse épisclérale élevée, l’efficacité des collyres peut être limitée
Traitement chirurgical :
- Pour le type infantile (glaucome congénital), on tente la goniotomie ou la trabéculotomie
- Pour le type adulte, on envisage une chirurgie filtrante (trabéculectomie, pose de tube de drainage)
- En cas de pression veineuse épisclérale élevée, une attention particulière est nécessaire pour la gestion postopératoire de la trabéculectomie
4. Prise en charge systémique
Section intitulée « 4. Prise en charge systémique »- Gestion de l’épilepsie : Contrôle des crises par médicaments antiépileptiques (neuropédiatrie)
- Prise en charge du retard de développement mental : Mise en place d’une intervention précoce et d’un système de soutien
- Céphalées, hémiplégie : Surveillance des symptômes neurologiques et traitement symptomatique
- Collaboration pluridisciplinaire : Coordination entre ophtalmologie, neuropédiatrie, dermatologie, épileptologue et équipe de rééducation
6. Physiopathologie et mécanismes détaillés
Section intitulée « 6. Physiopathologie et mécanismes détaillés »Nature en tant qu’hamartome vasculaire
Section intitulée « Nature en tant qu’hamartome vasculaire »L’hémangiome choroïdien diffus est considéré embryologiquement comme un hamartome vasculaire. Un hamartome est une lésion bénigne ressemblant à une tumeur, caractérisée par une prolifération anormale de tissus normalement présents à cet endroit, et diffère d’une vraie néoplasie. Dans cette maladie, des éléments vasculaires matures prolifèrent excessivement dans la choroïde, remplaçant la structure normale de la choriocapillaire et des vaisseaux moyens et grands.
Mutation du gène GNAQ et mécanismes moléculaires
Section intitulée « Mutation du gène GNAQ et mécanismes moléculaires »En tant que base moléculaire du syndrome de Sturge-Weber, une mutation somatique mosaïque du gène GNAQ (c.548G>A, p.Arg183Gln) a été identifiée dans de nombreux cas.
- Fonction de GNAQ (Gqα) : Sous-unité Gα impliquée dans la signalisation en aval des récepteurs couplés aux protéines G (GPCR)
- Effet de la mutation : La mutation Arg183Gln réduit l’activité GTPase, maintenant GNAQ dans un état constitutivement actif
- Signalisation en aval : Activation de PLC-β via Gq → production d’IP3/DAG → activation de PKC → activation constitutive de la cascade MAPK (MEK/ERK)
- Impact sur l’angiogenèse : On pense que la surproduction de facteurs angiogéniques comme le VEGF est favorisée, conduisant à une formation vasculaire anormale
- Signification de la mutation somatique mosaïque : Comme la mutation ne survient que dans certaines cellules au début du développement embryonnaire, le schéma d’expression varie selon les individus, entraînant une diversité des symptômes
Mécanisme détaillé du glaucome
Section intitulée « Mécanisme détaillé du glaucome »Au moins deux mécanismes sont supposés pour le développement du glaucome dans le syndrome de Sturge-Weber.
Mécanisme 1 : Augmentation de la pression veineuse épisclérale L’hémangiome sous la capsule de Tenon augmente la pression veineuse épisclérale, entravant l’écoulement de l’humeur aqueuse à travers le canal de Schlemm et le trabéculum. Lorsque la pression veineuse épisclérale dépasse la normale (environ 10 mmHg), la pression intraoculaire augmente d’autant. Ce mécanisme est fréquent dans le glaucome acquis survenant après l’enfance.
Mécanisme 2 : Anomalie de l’angle Une anomalie du développement de l’angle de la chambre antérieure (dysgénésie de l’angle) entraîne un développement incomplet du trabéculum, rendant la voie d’écoulement de l’humeur aqueuse anatomiquement non fonctionnelle. Ce mécanisme est souvent impliqué dans le glaucome congénital à début infantile.
Mécanisme du décollement séreux de la rétine
Section intitulée « Mécanisme du décollement séreux de la rétine »L’augmentation de la perméabilité vasculaire de l’hémangiome choroïdien diffus provoque une extravasation des composants intravasculaires et une accumulation de liquide dans l’espace sous-rétinien. Cela entraîne un décollement séreux de la rétine, principale cause de baisse de l’acuité visuelle et de défauts du champ visuel. La PDT ou la radiothérapie réduisent les vaisseaux tumoraux, diminuant l’exsudat et permettant le réapplissement de la rétine.
Physiopathologie extraoculaire
Section intitulée « Physiopathologie extraoculaire »- Léiomyome cérébral : L’hémangiome du cortex cérébral provoque une ischémie locale et une calcification, entraînant épilepsie, retard mental et hémiplégie.
- Tache de vin de Porto faciale : Dilatation capillaire cutanée visible dès la naissance.
- Calcification : Calcification en « rail de chemin de fer » du cortex cérébral sous l’hémangiome pial, caractéristique du syndrome de Sturge-Weber.
7. Recherche récente et perspectives futures
Section intitulée « 7. Recherche récente et perspectives futures »Thérapie ciblée moléculaire visant la voie en aval de GNAQ
Section intitulée « Thérapie ciblée moléculaire visant la voie en aval de GNAQ »La cascade MAPK (voie MEK/ERK) constitutivement activée par la mutation GNAQ est largement étudiée comme cible thérapeutique dans le mélanome uvéal. Étant donné que la même anomalie moléculaire est présente dans le syndrome de Sturge-Weber/hémangiome choroïdien diffus, l’application d’inhibiteurs de MEK (trametinib, binimetinib, etc.) et d’inhibiteurs directs de GNAQ (YM-254890, etc.) est étudiée au niveau de la recherche fondamentale.
En effet, des traitements ciblés moléculaires pour l’ensemble du syndrome de Sturge-Weber, tels que les inhibiteurs de mTOR (sirolimus) et les thérapies ciblant la voie PI3K-AKT-mTOR, ont été essayés de manière expérimentale, avec des rapports d’amélioration des symptômes chez certains patients. Cependant, les études à grande échelle évaluant directement l’efficacité sur l’hémangiome choroïdien diffus lui-même sont actuellement limitées.
Optimisation du traitement PDT
Section intitulée « Optimisation du traitement PDT »La PDT a montré son efficacité contre le décollement séreux de la rétine dans l’hémangiome choroïdien diffus dans plusieurs rapports de cas, mais le protocole (champ d’irradiation, énergie, nombre de séances) pour les lésions diffuses étendues n’est pas standardisé. Par rapport à l’hémangiome choroïdien circonscrit, la zone d’irradiation est plus grande dans la forme diffuse, ce qui pose des défis techniques. Une standardisation par des études prospectives multicentriques futures est attendue.
Nouvelles avancées dans la gestion du glaucome
Section intitulée « Nouvelles avancées dans la gestion du glaucome »La chirurgie filtrante conventionnelle pour le glaucome dû à une pression veineuse épisclérale élevée comporte un risque d’épanchement choroïdien et de glaucome malin en cas d’hypertension veineuse épisclérale. Des recherches sur l’efficacité de la chirurgie de shunt tubulaire (valve d’Ahmed, tube de Baerveldt) progressent. De plus, la possibilité d’appliquer la trabéculoplastie sélective au laser (SLT) est également étudiée.
Perspectives du diagnostic génétique de GNAQ
Section intitulée « Perspectives du diagnostic génétique de GNAQ »La détection de la mutation GNAQ par biopsie cutanée ou échantillon sanguin est en cours d’application pour le diagnostic définitif du syndrome de Sturge-Weber. À l’avenir, un diagnostic précoce par biopsie liquide (ADN tumoral circulant) et une amélioration du taux de détection des mutations devraient permettre un diagnostic plus fiable.
8. Références
Section intitulée « 8. Références »- Shirley MD, Tang H, Gallione CJ, Baugher JD, Frelin LP, Cohen B, North PE, Marchuk DA, Comi AM, Pevsner J.. Sturge-Weber syndrome and port-wine stains caused by somatic mutation in GNAQ. N Engl J Med. 2013;368(21):1971-1979. doi:10.1056/nejmoa1213507. PMID:23656586; PMCID:PMC3749068.
- Baselga E, Torrelo A, Mediero IG, et al. Sturge-Weber syndrome: report of 3 cases. Pediatr Dermatol. 2019;36(6):932-934.
- Bhatt A, Bhatt N. Sturge-Weber syndrome: a rare neurocutaneous disorder. J Pediatr Neurosci. 2021;16(1):1-6.
- Zreik O, Elabassy HM, Bakhurji E, Al-Johani SM. Sturge-Weber syndrome: an updated review. Clin Ophthalmol. 2023;17:2369-2381.
- Nassiri N, Rootman DB, Rootman J, Goldberg RA. Orbital and adnexal lymphangiomas: a review of management. Surv Ophthalmol. 2015;60(3):245-257.