Sporadique (le plus fréquent)
Mode de transmission : non héréditaire (sporadique)
Distribution des lésions : unilatéral, rétine périphérique
Évolution clinique : généralement non progressive, asymptomatique
Complications systémiques : aucune

L’hémangiome caverneux rétinien (retinal cavernous hemangioma) est une malformation vasculaire rétinienne formée par un regroupement de veines dilatées à faible débit. Généralement non héréditaire, unilatéral et non progressif, il survient de manière sporadique dans la rétine périphérique. Des cas avec atteinte de la tête du nerf optique ou de la macula, ainsi que des formes autosomiques dominantes associant des hémangiomes cutanés, cérébraux et hépatiques, ont été rapportés.
Cette maladie n’est pas strictement une tumeur mais une malformation vasculaire. Il s’agit d’un amas de vaisseaux composé de cellules endothéliales, de cellules musculaires lisses et de cellules stromales, sans prolifération monoclonale. Selon la classification de la Société internationale pour l’étude des anomalies vasculaires (ISSVA), elle est classée comme malformation veineuse à faible débit (venous malformation) et fait partie des maladies couvertes par les directives pour le diagnostic et le traitement des hémangiomes et malformations vasculaires.
Sporadique (le plus fréquent)
Mode de transmission : non héréditaire (sporadique)
Distribution des lésions : unilatéral, rétine périphérique
Évolution clinique : généralement non progressive, asymptomatique
Complications systémiques : aucune
Familial (autosomique dominant)
Mode de transmission : autosomique dominant
Distribution des lésions : unilatéral ou bilatéral, multiples
Évolution clinique : complications cérébrales, cutanées et hépatiques possibles
Complications systémiques : cavernome cérébral (CCM), angiome cutané, angiome hépatique
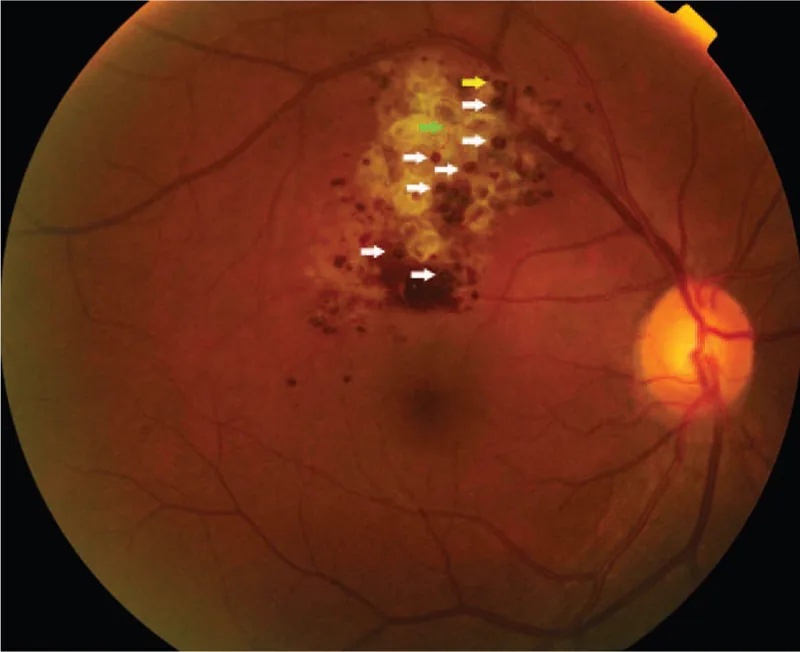
Lorsque la lésion est limitée à la périphérie, elle est généralement asymptomatique et souvent découverte fortuitement lors d’un examen du fond d’œil. Si la lésion s’étend à la papille optique ou à la macula, une baisse de l’acuité visuelle et un déficit du champ visuel peuvent survenir. Lorsqu’une membrane fibroproliférative se forme sur la tumeur et provoque une exsudation ou une hémorragie par traction, l’impact sur la fonction visuelle devient évident.
Les signes du fond d’œil sont les plus importants pour le diagnostic de cette maladie. Les signes caractéristiques sont présentés ci-dessous.
L’angiographie à la fluorescéine est l’examen le plus utile pour le diagnostic différentiel. Les signes caractéristiques sont présentés ci-dessous.
En tomographie par cohérence optique (OCT), la lésion apparaît comme une masse kystique hyperréflective en relief faisant saillie à partir des couches internes de la rétine. L’angiographie OCT (OCT-A) montre un flux sanguin interne faible, confirmant les caractéristiques d’une malformation vasculaire à faible débit.
Le mécanisme exact de l’hémangiome caverneux rétinien n’est pas entièrement élucidé. On pense qu’il est dû à une anomalie du développement vasculaire embryonnaire, à savoir un changement hamartomateux localisé du système veineux. Aucun facteur de risque environnemental pour les cas sporadiques n’a été identifié à ce jour.
Dans les cas familiaux (transmission autosomique dominante), il existe un fond génétique commun avec les cavernomes cérébraux (malformation caverneuse cérébrale, CCM). Trois gènes responsables de la CCM ont été identifiés, et leur association avec des malformations vasculaires systémiques, y compris les lésions oculaires, a été rapportée1).
| Gène | Alias | Protéine codée | Fonction principale |
|---|---|---|---|
| CCM1 | KRIT1 | Krev interaction trapped 1 | Adhésion cellulaire et homéostasie endothéliale vasculaire |
| CCM2 | MGC4607 | Malcavernine | Liaison à CCM1 et transduction du signal |
| CCM3 | PDCD10 | Programmed cell death 10 | Contrôle de l’apoptose et de la perméabilité vasculaire |
Ces mutations génétiques affectent l’adhésion cellulaire, la signalisation et le contrôle de la perméabilité des cellules endothéliales vasculaires, entraînant une dilatation veineuse et une fragilité de la paroi vasculaire2).
Les données précises sur l’incidence sont limitées, mais l’hémangiome caverneux rétinien est considéré comme une maladie rare3). La prévalence de l’hémangiome caverneux cérébral dans la population générale est de 0,1 à 0,5 %, mais la proportion de patients présentant des lésions oculaires est encore plus faible. Il existe peu de données claires sur l’âge de prédilection et le sexe. Les cas familiaux sont rares, mais plusieurs familles avec des associations d’hémangiomes cérébraux, cutanés et hépatiques ont été rapportées.
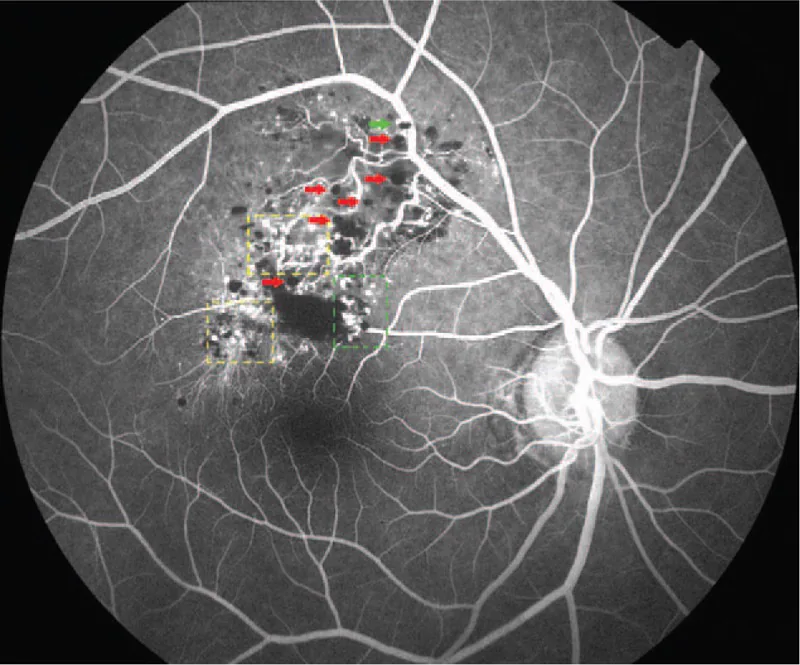
Le diagnostic repose principalement sur les signes caractéristiques du fond d’œil (tumeur multiloculaire rouge foncé en grappe de raisin) et les résultats de l’angiographie fluorescéinique (signe du capuchon fluorescent, motif de remplissage veineux avec peu de fuite de fluorescence). Dans les cas typiques, une biopsie n’est pas nécessaire et un diagnostic clinique est possible.
Dans les cas familiaux (hérédité autosomique dominante) d’angiome caverneux rétinien, il existe une association génétique avec l’angiome caverneux cérébral (CCM). Les mutations des gènes CCM1, CCM2 et CCM3 sont une cause commune, entraînant des malformations vasculaires multiples dans la rétine, le cerveau, la peau et le foie. Dans les cas familiaux, une IRM cérébrale avec contraste est obligatoire pour exclure des lésions cérébrales non découvertes (pouvant provoquer épilepsie ou hémorragie cérébrale). Dans les cas sporadiques, aucun lien avec des lésions cérébrales n’a été démontré.
Le diagnostic différentiel le plus important de l’angiome caverneux rétinien est l’hémangioblastome rétinien (maladie de von Hippel-Lindau). Les résultats de l’angiographie à la fluorescéine sont le point clé pour distinguer les deux maladies.
| Maladie | Aspect du fond d’œil | Aspect à l’angiographie à la fluorescéine (FA) | Complications systémiques | Traitement |
|---|---|---|---|---|
| Angiome caverneux rétinien | Tumeur rouge foncé multilobée (en grappe de raisin) | Remplissage veineux lent, signe du capuchon fluorescent, peu de fuite | CCM (familial) | Surveillance habituelle |
| Hémangiome capillaire rétinien (maladie de VHL) | Tumeur semi-transparente orange-rouge + vaisseaux afférents et efférents dilatés | Remplissage rapide en phase artérielle, fuite fluorescente abondante | Tumeur VHL (carcinome rénal, etc.) | Laser, chirurgie, anti-VEGF |
| Maladie de Coats | Décollement de rétine exsudatif, télangiectasies capillaires | Fuite abondante à partir des télangiectasies | Aucune (surtout chez l’enfant) | Laser, chirurgie |
| Tumeur rétinienne proliférante vasculaire | Masse jaune-blanc périphérique | Remplissage et fuite progressifs | Aucune | Laser, PDT |
Les deux maladies sont des lésions vasculaires rétiniennes, mais elles peuvent être clairement distinguées par l’angiographie à la fluorescéine. L’hémangiome capillaire rétinien (maladie de VHL) se caractérise par une tumeur orange-rouge avec des vaisseaux afférents et efférents dilatés, et l’angiographie montre un remplissage rapide dès la phase artérielle avec une fuite fluorescente abondante. En revanche, l’hémangiome caverneux rétinien se caractérise par un remplissage lent pendant la phase veineuse et un signe de calotte fluorescente, avec peu de fuite fluorescente même tardivement. À l’examen du fond d’œil, l’hémangiome caverneux rétinien apparaît comme une masse multiloculaire rouge foncé en grappe de raisin, ce qui permet généralement de le distinguer.
L’hémangiome caverneux rétinien est non progressif et ne nécessite généralement pas de traitement. Une membrane fibroproliférative peut se former sur la lésion, pouvant entraîner une exsudation ou une hémorragie par traction. Chez les patients asymptomatiques sans complications, une surveillance régulière par examen du fond d’œil est recommandée.
Un traitement est envisagé en cas de complications suivantes.
Vitrectomie : En cas de décollement de rétine par traction dû à une membrane fibroproliférative, ou d’hémorragie vitréenne associée à une traction, une vitrectomie peut être envisagée. Comme il s’agit d’une malformation vasculaire à faible débit, le risque hémorragique per- et postopératoire est considéré comme similaire à celui de la chirurgie rétinienne standard. La plupart des données sur les résultats chirurgicaux proviennent de rapports de cas, et les preuves à grande échelle sont limitées4).
Photocoagulation au laser : Étant donné qu’il s’agit d’une malformation vasculaire à faible débit, la réponse à la photocoagulation est faible. À l’heure actuelle, elle n’est pas activement recommandée.
Dans les cas familiaux, en plus de la gestion des lésions oculaires, une prise en charge neurologique pour les cavernomes cérébraux est importante. Pour les lésions cérébrales asymptomatiques, la surveillance est la règle, mais en cas de crises d’épilepsie ou de symptômes neurologiques, un traitement médicamenteux ou une intervention chirurgicale sont envisagés5).
En général, aucun traitement n’est nécessaire. Il s’agit d’une malformation vasculaire non progressive ; chez les patients asymptomatiques, la surveillance par examen du fond d’œil régulier est la règle. Ce n’est qu’en cas de complications tractionnelles dues à une membrane fibroproliférative (décollement de rétine par traction, hémorragie du vitré) que la vitrectomie est indiquée. La photocoagulation au laser est peu efficace en raison du faible débit et n’est généralement pas pratiquée. Dans les cas familiaux, en plus du suivi ophtalmologique, une prise en charge systémique des angiomes cérébraux est nécessaire.
L’hémangiome caverneux rétinien n’est pas une tumeur mais une malformation vasculaire. Il s’agit d’un amas de vaisseaux composé de cellules endothéliales, de cellules musculaires lisses et de cellules stromales, et non d’une prolifération monoclonale (prolifération tumorale). Dans la classification révisée de l’ISSVA (Société Internationale pour l’Étude des Anomalies Vasculaires) de 2018, il est classé comme malformation veineuse à faible débit, distincte des malformations artérioveineuses (haut débit)6).
Dans les cas familiaux (transmission autosomique dominante), les mutations avec perte de fonction des gènes CCM jouent un rôle central. CCM1 (KRIT1) régule la voie des intégrines impliquée dans l’adhésion intercellulaire, CCM2 (malcavernine) sert de protéine d’échafaudage pour CCM1, et CCM3 (PDCD10) est impliqué dans la régulation de l’apoptose et de la perméabilité vasculaire2). Ces mutations entraînent une rupture de l’adhésion intercellulaire des cellules endothéliales, une transition endothélio-mésenchymateuse, une dilatation vasculaire et une augmentation de la perméabilité.
Dans les cas sporadiques (non héréditaires), une anomalie locale du développement vasculaire due à une mutation somatique est suggérée. On pense qu’une anomalie au cours du développement vasculaire rétinien embryonnaire forme des dilatations veineuses polykystiques en grappe de raisin, mais les mécanismes détaillés restent largement inconnus.
Le fait qu’il s’agisse d’une malformation veineuse à faible débit détermine les caractéristiques cliniques de cette maladie. En raison du faible débit sanguin, les caractéristiques suivantes apparaissent :
Une prolifération gliale (gliotic cap) ou une membrane fibroproliférative peut se former à la surface (côté vitréen) de la malformation vasculaire. La contraction de cette membrane peut provoquer un décollement de rétine tractionnel ou une hémorragie du vitré. La membrane fibroproliférative n’est pas tumorale mais une prolifération réactive secondaire.
L’élucidation des mécanismes moléculaires liés aux mutations du gène CCM progresse. Dans les études animales, les inhibiteurs de la signalisation Rho/ROCK et les inhibiteurs de la voie PI3K/Akt/mTOR ont montré des résultats prometteurs dans des modèles d’angiomes caverneux cérébraux7). Des recherches supplémentaires sont nécessaires pour l’application aux lésions oculaires.
Les tests multi-gènes par séquençage de nouvelle génération (NGS) se sont généralisés et sont utilisés pour le diagnostic définitif des cas familiaux, le diagnostic des porteurs et le conseil génétique familial. L’interprétation des variants pathogènes des mutations du gène CCM s’accumule également1).
En ce qui concerne la gestion des complications tractionnelles par vitrectomie, la littérature sous forme de rapports de cas et de séries de cas s’accumule4,8). Il n’existe pas d’essai contrôlé randomisé à grande échelle, et des études prospectives futures sont souhaitées.
Fischer A, Zalvide J, Faurobert E, Albiges-Rizo C, Tournier-Lasserve E.. Cerebral cavernous malformations: from CCM genes to endothelial cell homeostasis. Trends Mol Med. 2013;19(5):302-308. doi:10.1016/j.molmed.2013.02.004. PMID:23506982.
Couteulx SL, Jung HH, Labauge P, et al. Truncating mutations in CCM1, encoding KRIT1, cause hereditary cavernous angiomas. Nat Genet. 1999;23(2):189-193. doi:10.1038/13815.
Gass JDM. Cavernous hemangioma of the retina. A neuro-oculocutaneous syndrome. Am J Ophthalmol. 1971;71(4):799-814.
Messmer E, Font RL, Laqua H, Höpping W, Naumann GO. Cavernous hemangioma of the retina. Immunohistochemical and ultrastructural observations. Archives of ophthalmology (Chicago, Ill. : 1960). 1984;102(3):413-8. doi:10.1001/archopht.1984.01040030331031. PMID:6538410.
Haller JA Jr, Dortz J, Goldberg MF. Familial retinal cavernous hemangiomas. Arch Ophthalmol. 1979;97(5):879-883.
ISSVA Classification of Vascular Anomalies. International Society for the Study of Vascular Anomalies. 2018 update. Available at: https://www.issva.org.
Lewis RA, Cohen BH, Wise GN. Cavernous haemangioma of the retina and optic disc. A report of three cases and a review of the literature. Br J Ophthalmol. 1975;59(8):422-434.
Shields JA, Shields CL, Timmers E, et al. Spectrum of vitreoretinal surgery. Retina. 1992;12(1):1-11.