Sporadisch (am häufigsten)
Vererbungsmuster : nicht erblich (sporadisch)
Läsionsverteilung : einseitig, periphere Netzhaut
Klinischer Verlauf : in der Regel nicht fortschreitend, asymptomatisch
Systemische Komplikationen : keine

Das retinale kavernöse Hämangiom (retinal cavernous hemangioma) ist eine Gefäßfehlbildung der Netzhaut, die aus einer Ansammlung erweiterter Venen mit niedrigem Fluss besteht. In der Regel ist es nicht erblich, einseitig und nicht fortschreitend und tritt sporadisch in der peripheren Netzhaut auf. Es wurden auch Fälle mit Beteiligung der Papille oder Makula sowie autosomal-dominante Formen mit Haut-, ZNS- und Leberhämangiomen berichtet.
Diese Erkrankung ist streng genommen kein Tumor, sondern eine Gefäßfehlbildung. Es handelt sich um eine Ansammlung von Gefäßen, die aus Endothelzellen, glatten Muskelzellen und Stromazellen besteht, ohne monoklonale Zellproliferation. Nach der Klassifikation der International Society for the Study of Vascular Anomalies (ISSVA) wird sie als venöse Malformation mit niedrigem Fluss (venous malformation) eingestuft und ist eine der Erkrankungen, die in den Leitlinien zur Diagnose und Behandlung von Hämangiomen und Gefäßfehlbildungen aufgeführt sind.
Sporadisch (am häufigsten)
Vererbungsmuster : nicht erblich (sporadisch)
Läsionsverteilung : einseitig, periphere Netzhaut
Klinischer Verlauf : in der Regel nicht fortschreitend, asymptomatisch
Systemische Komplikationen : keine
Familiär (autosomal-dominant)
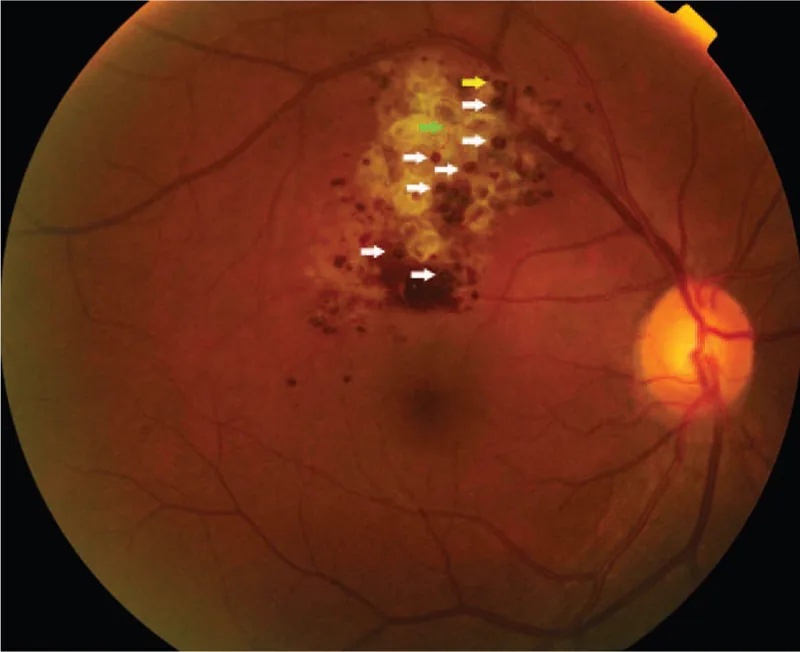
Wenn die Läsion auf die Peripherie beschränkt ist, ist sie in der Regel asymptomatisch und wird oft zufällig bei einer Fundusuntersuchung entdeckt. Wenn die Läsion die Papille oder Makula betrifft, können Sehstörungen und Gesichtsfeldausfälle auftreten. Wenn sich auf dem Tumor eine fibröse Proliferationsmembran bildet und durch Zug Exsudation oder Blutung verursacht wird, wird die Beeinträchtigung der Sehfunktion offensichtlich.
Die Fundusbefunde sind für die Diagnose dieser Erkrankung am wichtigsten. Charakteristische Befunde sind im Folgenden aufgeführt.
Die Fluoreszenzangiographie ist die nützlichste Untersuchung für die Differentialdiagnose. Charakteristische Befunde sind im Folgenden aufgeführt.
In der optischen Kohärenztomographie (OCT) stellt sich die Läsion als zystische, erhabene, hyperreflektive Raumforderung dar, die aus den inneren Netzhautschichten hervorragt. Die OCT-Angiographie (OCT-A) zeigt einen schwachen inneren Blutfluss und bestätigt damit die Merkmale einer Niedrigfluss-Gefäßfehlbildung.
Der genaue Entstehungsmechanismus des retinalen kavernösen Hämangioms ist nicht vollständig geklärt. Es wird angenommen, dass es auf eine Anomalie der embryonalen Gefäßentwicklung zurückzuführen ist, nämlich eine lokalisierte hamartomatöse Veränderung des Venensystems. Für sporadische Fälle wurden bisher keine umweltbedingten Risikofaktoren identifiziert.
Bei familiären Fällen (autosomal-dominante Vererbung) besteht ein gemeinsamer genetischer Hintergrund mit zerebralen kavernösen Malformationen (CCM). Drei ursächliche Gene für CCM wurden identifiziert, und ihre Assoziation mit systemischen Gefäßmalformationen einschließlich Augenläsionen wurde berichtet1).
| Gen | Alias | Codiertes Protein | Hauptfunktion |
|---|---|---|---|
| CCM1 | KRIT1 | Krev interaction trapped 1 | Zelladhäsion und vaskuläre endotheliale Homöostase |
| CCM2 | MGC4607 | Malcavernin | Bindung an CCM1 und Signaltransduktion |
| CCM3 | PDCD10 | Programmed cell death 10 | Kontrolle von Apoptose und Gefäßpermeabilität |
Diese Genmutationen beeinflussen die Zelladhäsion, Signaltransduktion und Permeabilitätskontrolle von vaskulären Endothelzellen, was zu venöser Dilatation und Schwächung der Gefäßwand führt2).
Genaue Inzidenzdaten sind begrenzt, aber das retinale kavernöse Hämangiom gilt als seltene Erkrankung3). Die bevölkerungsbasierte Prävalenz des zerebralen kavernösen Hämangioms beträgt 0,1–0,5 %, aber der Anteil mit Augenläsionen ist noch geringer. Es gibt wenige klare Daten zu bevorzugtem Alter und Geschlecht. Familiäre Fälle sind selten, aber mehrere Familien mit Assoziation von zerebralen, kutanen und hepatischen Hämangiomen wurden berichtet.
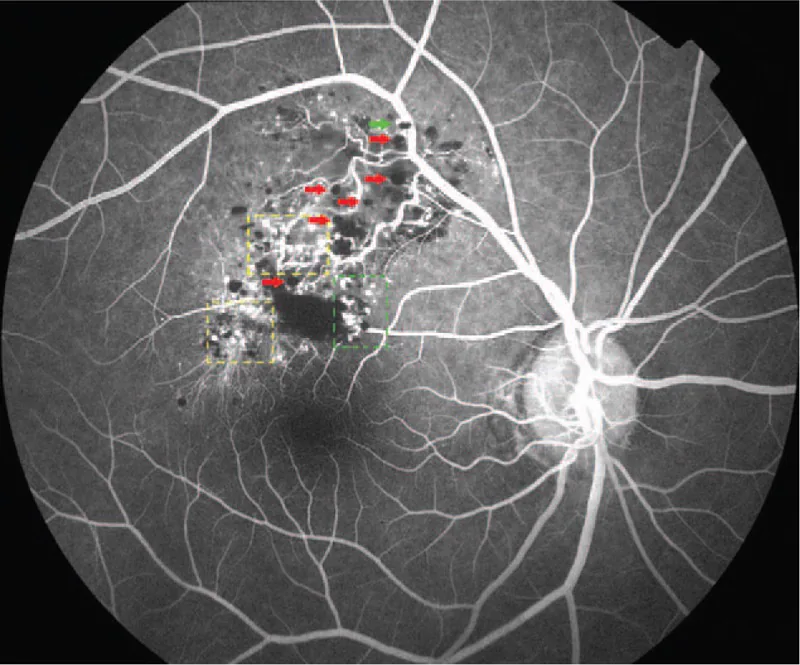
Die Diagnose basiert hauptsächlich auf den charakteristischen Fundusbefunden (traubenartiger, multilokulärer, dunkelroter Tumor) und den Fluoreszenzangiographie-Befunden (fluorescent cap sign, venöses Füllmuster mit geringem Fluoreszenzaustritt). In typischen Fällen ist keine Biopsie erforderlich, und eine klinische Diagnose ist möglich.
Bei familiären (autosomal-dominant vererbten) retinalen kavernösen Angiomen besteht ein genetischer Zusammenhang mit zerebralen kavernösen Angiomen (CCM). Mutationen in den Genen CCM1, CCM2 und CCM3 sind eine gemeinsame Ursache und führen zu multiplen vaskulären Fehlbildungen in Netzhaut, Gehirn, Haut und Leber. Bei familiären Fällen ist eine kontrastmittelverstärkte MRT des Gehirns obligatorisch, um unentdeckte Hirnläsionen (die Epilepsie oder Hirnblutungen verursachen können) auszuschließen. Bei sporadischen Fällen wurde kein Zusammenhang mit Hirnläsionen nachgewiesen.
Die wichtigste Differenzialdiagnose des retinalen kavernösen Angioms ist das retinale kapilläre Hämangiom (Morbus von Hippel-Lindau). Die Befunde der Fluoreszenzangiographie sind der entscheidende Punkt zur Unterscheidung beider Erkrankungen.
| Erkrankung | Fundusbefund | FA-Befund | Systemische Komplikationen | Behandlung |
|---|---|---|---|---|
| Retinales kavernöses Angiom | Multizystische dunkelrote Raumforderung (traubenartig) | Langsame venöse Füllung, fluoreszierendes Kappenzeichen, geringe Leckage | CCM (familiär) | In der Regel Beobachtung |
| Retinales kapilläres Hämangiom (VHL-Krankheit) | Orange-rote halbtransparente Tumormasse + erweiterte zu- und abführende Gefäße | Schnelle Füllung in der arteriellen Phase, starke Fluoreszein-Leckage | VHL-Tumor (Nierenzellkarzinom usw.) | Laser, Chirurgie, Anti-VEGF |
| Morbus Coats | Exsudative Netzhautablösung, kapilläre Teleangiektasien | Starke Leckage aus Teleangiektasien | Keine (meist Kinder) | Laser, Chirurgie |
| Retinaler vaskulärer proliferativer Tumor | Periphere gelb-weiße Raumforderung | Allmähliche Füllung und Leckage | Keine | Laser, PDT |
Beide Erkrankungen sind vaskuläre Läsionen der Netzhaut, können aber durch die Fluoreszenzangiographie klar unterschieden werden. Das retinale Kapillarhämangiom (Morbus VHL) ist gekennzeichnet durch einen orangeroten Tumor mit erweiterten zu- und abführenden Gefäßen; in der FA zeigt sich eine schnelle Füllung in der arteriellen Phase mit starkem Fluoreszenzaustritt. Das retinale kavernöse Hämangiom hingegen ist gekennzeichnet durch eine langsame Füllung in der venösen Phase und ein fluoreszierendes Kappenzeichen, mit nur geringem Fluoreszenzaustritt auch in späteren Phasen. Im Fundusbefund erscheint das retinale kavernöse Hämangiom als traubenartige, multilokuläre, dunkelrote Raumforderung, sodass eine Unterscheidung in der Regel möglich ist.
Das retinale kavernöse Hämangiom ist nicht progredient und erfordert in der Regel keine Behandlung. Auf der Läsion kann sich eine fibroproliferative Membran bilden, die durch Zug zu Exsudation oder Blutung führen kann. Bei asymptomatischen Patienten ohne Komplikationen erfolgt eine regelmäßige Kontrolle durch Fundusuntersuchung.
Eine Behandlung wird bei Auftreten folgender Komplikationen in Betracht gezogen.
Vitrektomie: Bei einer durch eine fibroproliferative Membran verursachten traktiven Netzhautablösung oder einer mit Traktion verbundenen Glaskörperblutung wird eine Vitrektomie erwogen. Da es sich um eine vaskuläre Fehlbildung mit niedrigem Fluss handelt, wird das Blutungsrisiko während und nach der Operation als ähnlich wie bei einer Standard-Netzhautchirurgie angesehen. Die meisten Daten zu Operationsergebnissen stammen aus Fallberichten, groß angelegte Evidenz ist rar4).
Laserphotokoagulation: Aufgrund der vaskulären Fehlbildung mit niedrigem Fluss ist die Reaktion auf die Photokoagulation gering. Derzeit wird sie nicht aktiv empfohlen.
Bei familiären Fällen ist neben der Behandlung der Augenveränderungen auch die neurologische Behandlung des zerebralen kavernösen Angioms wichtig. Bei asymptomatischen Hirnläsionen ist die Beobachtung die Grundlage, aber bei Auftreten von epileptischen Anfällen oder neurologischen Symptomen werden medikamentöse Therapie oder chirurgische Eingriffe in Betracht gezogen5).
In der Regel ist keine Behandlung erforderlich. Es handelt sich um eine nicht fortschreitende Gefäßfehlbildung; bei asymptomatischen Patienten ist die regelmäßige augenärztliche Kontrolle die Grundlage. Erst bei Auftreten von traktionsbedingten Komplikationen durch eine fibroproliferative Membran (Traktionsamotio, Glaskörperblutung) ist eine Vitrektomie indiziert. Die Laserphotokoagulation ist aufgrund des niedrigen Flusses wenig wirksam und wird in der Regel nicht durchgeführt. Bei familiären Fällen ist neben der augenärztlichen Nachsorge eine systemische Behandlung der zerebralen Angiome erforderlich.
Das retinale kavernöse Hämangiom ist kein Tumor, sondern eine Gefäßfehlbildung. Es handelt sich um eine Ansammlung von Gefäßen, die aus Endothelzellen, glatten Muskelzellen und Stromazellen besteht, und nicht um eine einzelne Zellproliferation (tumoröse Proliferation). In der überarbeiteten Klassifikation der ISSVA (International Society for the Study of Vascular Anomalies) von 2018 wird es als venöse Malformation mit niedrigem Fluss eingestuft, unterschieden von arteriovenösen Malformationen (hoher Fluss)6).
Bei familiären (autosomal-dominant vererbten) Fällen spielen Funktionsverlustmutationen der CCM-Gene eine zentrale Rolle. CCM1 (KRIT1) reguliert den Integrin-Signalweg, der an der Zell-Zell-Adhäsion beteiligt ist, CCM2 (Malcavernin) fungiert als Gerüstprotein für CCM1, und CCM3 (PDCD10) ist an der Apoptoseregulation und der Regulierung der Gefäßpermeabilität beteiligt2). Diese Mutationen führen zu einer Störung der Zell-Zell-Adhäsion der Endothelzellen, einer endothelial-mesenchymalen Transition, Gefäßerweiterung und erhöhter Permeabilität.
Bei sporadischen (nicht-hereditären) Fällen wird eine lokale Gefäßentwicklungsanomalie aufgrund einer somatischen Mutation vermutet. Es wird angenommen, dass eine Anomalie während der embryonalen Netzhautgefäßentwicklung zu traubenartigen, multizystischen Venenerweiterungen führt, aber die genauen Mechanismen sind noch weitgehend ungeklärt.
Die Tatsache, dass es sich um eine venöse Gefäßfehlbildung mit niedrigem Fluss handelt, bestimmt die klinischen Merkmale dieser Erkrankung. Aufgrund des geringen Blutflusses ergeben sich folgende Eigenschaften:
Auf der Oberfläche (Glaskörperseite) der Gefäßmissbildung kann sich eine gliöse Kappe oder eine fibroproliferative Membran bilden. Die Kontraktion dieser Membran kann zu einer traktiven Netzhautablösung oder Glaskörperblutung führen. Die fibroproliferative Membran ist nicht neoplastisch, sondern eine sekundäre reaktive Proliferation.
Die Aufklärung der molekularen Mechanismen durch CCM-Genmutationen schreitet voran. In Tierversuchen haben Rho/ROCK-Signalweg-Inhibitoren und PI3K/Akt/mTOR-Signalweg-Inhibitoren vielversprechende Ergebnisse in Modellen zerebraler kavernöser Angiome gezeigt7). Für die Anwendung bei Augenläsionen ist weitere Forschung erforderlich.
Multi-Gen-Panel-Tests mittels Next-Generation Sequencing (NGS) haben sich verbreitet und werden für die definitive Diagnose familiärer Fälle, die Trägerdiagnostik und die genetische Beratung von Familien genutzt. Auch die Interpretation pathogener Varianten von CCM-Genmutationen sammelt sich an1).
Zur Behandlung traktiver Komplikationen durch Vitrektomie sammelt sich Literatur auf dem Niveau von Fallberichten und Fallserien4,8). Es gibt keine großen randomisierten kontrollierten Studien, und zukünftige prospektive Studien sind wünschenswert.
Fischer A, Zalvide J, Faurobert E, Albiges-Rizo C, Tournier-Lasserve E.. Cerebral cavernous malformations: from CCM genes to endothelial cell homeostasis. Trends Mol Med. 2013;19(5):302-308. doi:10.1016/j.molmed.2013.02.004. PMID:23506982.
Couteulx SL, Jung HH, Labauge P, et al. Truncating mutations in CCM1, encoding KRIT1, cause hereditary cavernous angiomas. Nat Genet. 1999;23(2):189-193. doi:10.1038/13815.
Gass JDM. Cavernous hemangioma of the retina. A neuro-oculocutaneous syndrome. Am J Ophthalmol. 1971;71(4):799-814.
Messmer E, Font RL, Laqua H, Höpping W, Naumann GO. Cavernous hemangioma of the retina. Immunohistochemical and ultrastructural observations. Archives of ophthalmology (Chicago, Ill. : 1960). 1984;102(3):413-8. doi:10.1001/archopht.1984.01040030331031. PMID:6538410.
Haller JA Jr, Dortz J, Goldberg MF. Familial retinal cavernous hemangiomas. Arch Ophthalmol. 1979;97(5):879-883.
ISSVA Classification of Vascular Anomalies. International Society for the Study of Vascular Anomalies. 2018 update. Available at: https://www.issva.org.
Lewis RA, Cohen BH, Wise GN. Cavernous haemangioma of the retina and optic disc. A report of three cases and a review of the literature. Br J Ophthalmol. 1975;59(8):422-434.
Shields JA, Shields CL, Timmers E, et al. Spectrum of vitreoretinal surgery. Retina. 1992;12(1):1-11.