Thể lẻ tẻ (phổ biến nhất)
Kiểu di truyền: Không di truyền (lẻ tẻ)
Phân bố tổn thương: Một mắt, võng mạc ngoại vi
Diễn tiến lâm sàng: Thường không tiến triển, không triệu chứng
Biến chứng toàn thân: Không

U máu dạng hang võng mạc (retinal cavernous hemangioma) là một dị dạng mạch máu võng mạc bao gồm tập hợp các tĩnh mạch giãn lưu lượng thấp. Thường không di truyền, một mắt, không tiến triển, xảy ra đơn độc ở võng mạc ngoại vi. Đã có báo cáo về các trường hợp liên quan đến đĩa thị hoặc hoàng điểm, và các trường hợp di truyền trội kết hợp với dị dạng da, thần kinh trung ương và gan.
Bệnh này không phải là khối u theo nghĩa chặt chẽ, mà được phân loại là dị dạng mạch máu. Nó là một tập hợp mạch máu bao gồm tế bào nội mô, tế bào cơ trơn và tế bào mô đệm, không phải sự tăng sinh của một tế bào đơn lẻ. Theo phân loại của Hiệp hội Nghiên cứu Dị dạng Mạch máu Quốc tế (ISSVA), nó được xếp vào nhóm dị dạng tĩnh mạch lưu lượng thấp (venous malformation), và cũng là một bệnh trong hướng dẫn quản lý u máu và dị dạng mạch máu.
Thể lẻ tẻ (phổ biến nhất)
Kiểu di truyền: Không di truyền (lẻ tẻ)
Phân bố tổn thương: Một mắt, võng mạc ngoại vi
Diễn tiến lâm sàng: Thường không tiến triển, không triệu chứng
Biến chứng toàn thân: Không
Thể gia đình (di truyền trội nhiễm sắc thể thường)
Kiểu di truyền: Trội nhiễm sắc thể thường
Phân bố tổn thương: Một hoặc hai mắt, nhiều tổn thương
Diễn tiến lâm sàng: Có thể có biến chứng não, da, gan
Biến chứng toàn thân: U mạch hang não (CCM), u mạch máu da, u mạch máu gan
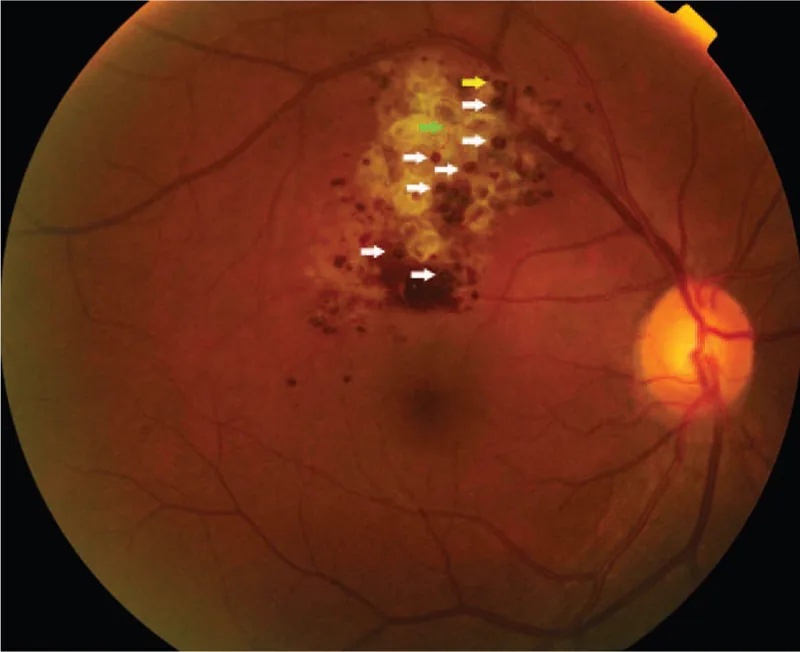
Khi tổn thương khu trú ở vùng ngoại vi, thường không có triệu chứng và thường được phát hiện tình cờ khi khám đáy mắt. Trong trường hợp tổn thương lan đến gai thị hoặc hoàng điểm, có thể xảy ra suy giảm thị lực và khiếm khuyết thị trường. Nếu màng xơ tăng sinh hình thành trên khối u và gây xuất tiết hoặc xuất huyết do co kéo, ảnh hưởng đến chức năng thị giác sẽ trở nên rõ rệt.
Hình ảnh đáy mắt là quan trọng nhất trong chẩn đoán bệnh này. Các đặc điểm điển hình được trình bày dưới đây.
Chụp mạch huỳnh quang là xét nghiệm hữu ích nhất để chẩn đoán phân biệt. Các đặc điểm điển hình được trình bày dưới đây.
Trên chụp cắt lớp quang học (OCT), tổn thương xuất hiện dưới dạng khối tăng âm dạng nang nhô ra từ các lớp võng mạc bên trong. Trên chụp mạch OCT (OCT-A), dòng máu bên trong tổn thương rất ít, xác nhận đặc điểm của nó như một dị dạng mạch máu lưu lượng thấp.
Cơ chế chính xác hình thành u máu hang võng mạc vẫn chưa được hiểu đầy đủ. Nguyên nhân được cho là do sự bất thường trong phát triển mạch máu thời kỳ phôi thai, tức là sự thay đổi dạng hamartoma cục bộ của hệ thống tĩnh mạch. Các yếu tố nguy cơ môi trường cho các trường hợp lẻ tẻ hiện chưa được xác định.
Trong các trường hợp gia đình (di truyền trội nhiễm sắc thể thường), có nền tảng di truyền chung với dị dạng hang não (CCM). Ba gen gây dị dạng hang não đã được xác định, và có báo cáo về mối liên quan của chúng với các dị dạng mạch máu toàn thân bao gồm tổn thương mắt1).
| Gen | Tên khác | Protein mã hóa | Chức năng chính |
|---|---|---|---|
| CCM1 | KRIT1 | Krev interaction trapped 1 | Kết dính tế bào và cân bằng nội môi nội mô mạch máu |
| CCM2 | MGC4607 | Malcavernin | Liên kết với CCM1 và truyền tín hiệu |
| CCM3 | PDCD10 | Chết tế bào theo chương trình 10 | Điều hòa quá trình chết tế bào và tính thấm mạch máu |
Các đột biến gen này liên quan đến sự kết dính tế bào nội mô mạch máu, truyền tín hiệu và điều hòa tính thấm, dẫn đến giãn tĩnh mạch và thành mạch yếu2).
Dữ liệu về tỷ lệ mắc chính xác còn hạn chế, nhưng u mạch hang võng mạc được coi là bệnh hiếm gặp3). Tỷ lệ hiện mắc u mạch hang não trong dân số là 0,1-0,5%, nhưng tỷ lệ có tổn thương mắt còn thấp hơn. Không có dữ liệu rõ ràng về độ tuổi hoặc giới tính thường gặp. Các trường hợp gia đình hiếm gặp, nhưng một số gia đình có kết hợp u mạch máu não, da và gan đã được báo cáo.
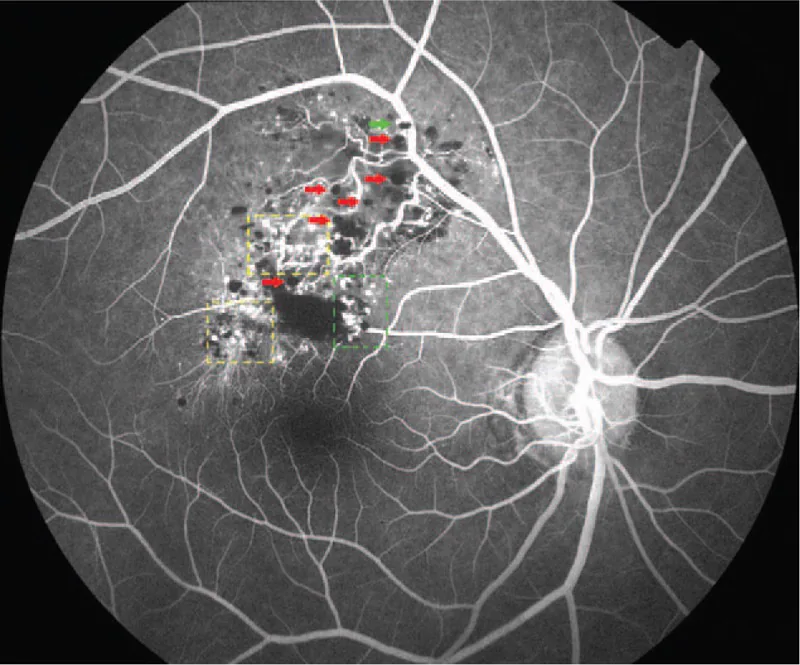
Chẩn đoán chủ yếu dựa trên các dấu hiệu đáy mắt đặc trưng (khối đa nang đỏ sẫm hình chùm nho) và kết quả chụp mạch huỳnh quang (dấu hiệu nắp huỳnh quang, kiểu nạp tĩnh mạch với rò rỉ huỳnh quang tối thiểu). Trong các trường hợp điển hình, không cần sinh thiết và có thể chẩn đoán lâm sàng.
Trong u hang mạch máu võng mạc gia đình (di truyền trội nhiễm sắc thể thường), có mối liên quan di truyền với u hang mạch máu não (CCM). Đột biến gen CCM1, CCM2 và CCM3 là nguyên nhân chung, gây ra các dị dạng mạch máu đa ổ ở võng mạc, não, da và gan. Ở các trường hợp gia đình, MRI não có tiêm thuốc cản quang là bắt buộc để loại trừ các tổn thương não chưa được phát hiện (có thể gây động kinh hoặc xuất huyết não). Ở các trường hợp lẻ tẻ, chưa thấy mối liên quan với tổn thương não.
Chẩn đoán phân biệt quan trọng nhất của u hang mạch máu võng mạc là u nguyên bào mạch máu võng mạc (bệnh VHL). Chụp mạch huỳnh quang là điểm quan trọng nhất để phân biệt hai bệnh.
| Bệnh | Hình ảnh đáy mắt | Hình ảnh FA | Biến chứng toàn thân | Điều trị |
|---|---|---|---|---|
| U hang mạch máu võng mạc | Khối đỏ sẫm nhiều thùy (giống chùm nho) | Đổ đầy tĩnh mạch chậm, dấu hiệu nắp huỳnh quang, ít thoát mạch | CCM (gia đình) | Thường theo dõi |
| U mạch mao mạch võng mạc (Bệnh VHL) | Khối u trong mờ màu đỏ cam kèm mạch máu đến và đi giãn rộng | Ngấm thuốc nhanh thì động mạch, thoát huỳnh quang mạnh | U VHL (ví dụ ung thư biểu mô tế bào thận) | Laser - phẫu thuật - kháng VEGF |
| Bệnh Coats | Bong võng mạc tiết dịch, phình mạch mao mạch | Thoát thuốc mạnh từ phình mạch mao mạch | Không có (thường gặp ở trẻ em) | Laser - phẫu thuật |
| U tăng sinh mạch máu võng mạc | Khối màu trắng vàng ở ngoại vi | Ngấm thuốc tăng dần, thoát thuốc | Không có | Laser - PDT |
Cả hai bệnh đều là tổn thương mạch máu võng mạc, nhưng có thể phân biệt rõ ràng bằng chụp mạch huỳnh quang. Trong u nguyên bào mạch máu võng mạc (bệnh VHL), có khối u màu đỏ cam kèm các mạch máu đến và đi giãn, trên FA, khối u ngấm thuốc nhanh từ thì động mạch với rò rỉ huỳnh quang mạnh. Ngược lại, u mạch hang võng mạc đặc trưng bởi ngấm thuốc chậm ở thì tĩnh mạch và dấu hiệu nắp huỳnh quang, rò rỉ huỳnh quang rất ít ngay cả ở thì muộn. Trên soi đáy mắt, u mạch hang võng mạc xuất hiện như một khối đa nang màu đỏ sẫm giống chùm nho, và thường có thể phân biệt được.
U mạch hang võng mạc không tiến triển và thường không cần điều trị. Màng tăng sinh xơ có thể hình thành trên tổn thương, có thể gây xuất tiết hoặc xuất huyết do co kéo. Ở những trường hợp không triệu chứng và không có biến chứng, tiến hành theo dõi định kỳ bằng khám đáy mắt.
Điều trị được xem xét khi có các biến chứng sau.
Phẫu thuật dịch kính: Trong trường hợp bong võng mạc do co kéo hoặc xuất huyết dịch kính do màng tăng sinh xơ, cân nhắc phẫu thuật dịch kính. Vì là dị dạng mạch máu lưu lượng thấp, nguy cơ chảy máu trong và sau phẫu thuật tương đương với phẫu thuật võng mạc thông thường. Hầu hết y văn là báo cáo ca bệnh, bằng chứng quy mô lớn còn hạn chế4).
Quang đông laser: Vì là dị dạng mạch máu lưu lượng thấp, đáp ứng với quang đông kém. Hiện tại không được khuyến cáo tích cực.
Trong các trường hợp gia đình, ngoài việc quản lý tổn thương mắt, quản lý thần kinh đối với u hang não là rất quan trọng. Đối với tổn thương não, nếu không có triệu chứng, theo dõi là cơ bản, nhưng nếu xuất hiện cơn động kinh hoặc triệu chứng thần kinh, cần xem xét điều trị bằng thuốc hoặc can thiệp phẫu thuật5).
Thông thường không cần điều trị. Đây là dị dạng mạch máu không tiến triển, và trong các trường hợp không triệu chứng, theo dõi định kỳ bằng khám đáy mắt là chính sách cơ bản. Chỉ khi có biến chứng kéo do màng tăng sinh xơ (bong võng mạc co kéo, xuất huyết dịch kính) thì phẫu thuật cắt dịch kính mới được chỉ định. Quang đông laser không hiệu quả do lưu lượng thấp và thường không được thực hiện. Trong các trường hợp gia đình, ngoài theo dõi nhãn khoa, cần quản lý toàn thân u hang não.
U hang võng mạc không phải là khối u mà là dị dạng mạch máu. Nó là một khối mạch máu bao gồm tế bào nội mô mạch máu, tế bào cơ trơn và tế bào mô kẽ, và không phải là sự tăng sinh của một tế bào đơn lẻ (tăng sinh tân sinh), đây là đặc điểm cốt lõi. Trong phân loại sửa đổi năm 2018 của ISSVA (Hiệp hội Nghiên cứu Dị dạng Mạch máu Quốc tế), nó được phân loại là dị dạng tĩnh mạch lưu lượng thấp (venous malformation), và được phân biệt với dị dạng động tĩnh mạch (lưu lượng cao)6).
Trong các trường hợp gia đình (di truyền trội nhiễm sắc thể thường), các đột biến mất chức năng của gen CCM đóng vai trò trung tâm. CCM1 (KRIT1) điều chỉnh con đường integrin liên quan đến kết dính gian bào, CCM2 (malcavernin) hoạt động như protein khung cho CCM1. CCM3 (PDCD10) tham gia vào điều hòa apoptosis và tính thấm mạch máu2). Các đột biến này gây ra phá vỡ kết dính gian bào của tế bào nội mô mạch máu → chuyển đổi nội mô-trung mô (endothelial-to-mesenchymal transition) → giãn mạch và tăng tính thấm.
Trong các trường hợp lẻ tẻ (không di truyền), nghi ngờ có bất thường phát triển mạch máu cục bộ do đột biến soma (somatic mutation). Người ta cho rằng bất thường trong quá trình tạo mạch võng mạc phôi thai hình thành các giãn tĩnh mạch đa nang giống chùm nho, nhưng cơ chế chi tiết vẫn chưa được làm sáng tỏ.
Là dị dạng mạch máu tĩnh mạch lưu lượng thấp quyết định các đặc điểm lâm sàng của bệnh này. Do lưu lượng máu thấp, các đặc điểm sau xuất hiện:
Trên bề mặt của dị dạng mạch máu (phía thủy tinh thể) có thể hình thành nắp thần kinh đệm (gliotic cap) hoặc màng tăng sinh xơ. Sự co rút của màng này có thể gây bong võng mạc co kéo hoặc xuất huyết dịch kính. Màng tăng sinh xơ không phải là khối u, mà là sự tăng sinh phản ứng thứ phát.
Sự hiểu biết về cơ chế phân tử do đột biến gen CCM đang tiến triển. Trong các thí nghiệm trên động vật, các chất ức chế tín hiệu Rho/ROCK và chất ức chế con đường PI3K/Akt/mTOR đã cho thấy kết quả khả quan trên mô hình u mạch hang não 7). Việc ứng dụng vào tổn thương mắt cần nghiên cứu thêm.
Xét nghiệm bảng gen đa gen bằng giải trình tự thế hệ mới (NGS) đã trở nên phổ biến, được sử dụng để chẩn đoán xác định các trường hợp gia đình, chẩn đoán người mang gen và tư vấn di truyền cho gia đình. Việc giải thích các biến thể bệnh lý của đột biến gen CCM cũng đang được tích lũy 1).
Về việc quản lý các biến chứng co kéo bằng phẫu thuật dịch kính, các tài liệu báo cáo ca bệnh và loạt ca bệnh đang được tích lũy 4,8). Chưa có thử nghiệm ngẫu nhiên có đối chứng quy mô lớn, và các nghiên cứu tiến cứu trong tương lai được mong đợi.
Fischer A, Zalvide J, Faurobert E, Albiges-Rizo C, Tournier-Lasserve E.. Cerebral cavernous malformations: from CCM genes to endothelial cell homeostasis. Trends Mol Med. 2013;19(5):302-308. doi:10.1016/j.molmed.2013.02.004. PMID:23506982.
Couteulx SL, Jung HH, Labauge P, et al. Truncating mutations in CCM1, encoding KRIT1, cause hereditary cavernous angiomas. Nat Genet. 1999;23(2):189-193. doi:10.1038/13815.
Gass JDM. Cavernous hemangioma of the retina. A neuro-oculocutaneous syndrome. Am J Ophthalmol. 1971;71(4):799-814.
Messmer E, Font RL, Laqua H, Höpping W, Naumann GO. Cavernous hemangioma of the retina. Immunohistochemical and ultrastructural observations. Archives of ophthalmology (Chicago, Ill. : 1960). 1984;102(3):413-8. doi:10.1001/archopht.1984.01040030331031. PMID:6538410.
Haller JA Jr, Dortz J, Goldberg MF. Familial retinal cavernous hemangiomas. Arch Ophthalmol. 1979;97(5):879-883.
ISSVA Classification of Vascular Anomalies. International Society for the Study of Vascular Anomalies. 2018 update. Available at: https://www.issva.org.
Lewis RA, Cohen BH, Wise GN. Cavernous haemangioma of the retina and optic disc. A report of three cases and a review of the literature. Br J Ophthalmol. 1975;59(8):422-434.
Shields JA, Shields CL, Timmers E, et al. Spectrum of vitreoretinal surgery. Retina. 1992;12(1):1-11.