Sporadico (più comune)
Modalità di trasmissione : non ereditaria (sporadica)
Distribuzione delle lesioni : unilaterale, retina periferica
Decorso clinico : solitamente non progressivo, asintomatico
Complicanze sistemiche : nessuna

L’emangioma cavernoso retinico (retinal cavernous hemangioma) è una malformazione vascolare della retina formata da un agglomerato di vene dilatate a basso flusso. Di solito è non ereditario, unilaterale e non progressivo, e si verifica sporadicamente nella retina periferica. Sono stati riportati anche casi con coinvolgimento della testa del nervo ottico o della macula, nonché forme autosomiche dominanti associate a emangiomi cutanei, cerebrali ed epatici.
Questa malattia non è strettamente un tumore ma una malformazione vascolare. Si tratta di un ammasso di vasi composto da cellule endoteliali, cellule muscolari lisce e cellule stromali, senza proliferazione monoclonale. Secondo la classificazione della International Society for the Study of Vascular Anomalies (ISSVA), è classificata come malformazione venosa a basso flusso (venous malformation) ed è una delle malattie coperte dalle linee guida per la diagnosi e il trattamento degli emangiomi e delle malformazioni vascolari.
Sporadico (più comune)
Modalità di trasmissione : non ereditaria (sporadica)
Distribuzione delle lesioni : unilaterale, retina periferica
Decorso clinico : solitamente non progressivo, asintomatico
Complicanze sistemiche : nessuna
Familiare (autosomico dominante)
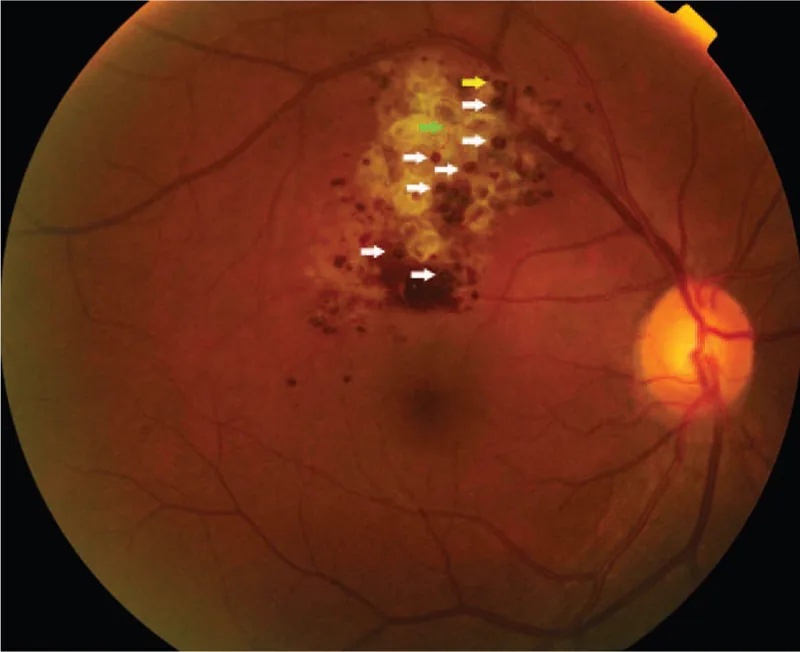
Quando la lesione è limitata alla periferia, di solito è asintomatica e spesso viene scoperta incidentalmente durante un esame del fondo oculare. Se la lesione si estende alla papilla ottica o alla macula, possono verificarsi diminuzione dell’acuità visiva e difetti del campo visivo. Quando sulla lesione si forma una membrana fibroproliferativa e si verificano essudazione o emorragia per trazione, l’impatto sulla funzione visiva diventa evidente.
I reperti del fondo oculare sono i più importanti per la diagnosi di questa malattia. I reperti caratteristici sono elencati di seguito.
L’angiografia con fluoresceina è l’esame più utile per la diagnosi differenziale. I reperti caratteristici sono elencati di seguito.
Alla tomografia a coerenza ottica (OCT), la lesione appare come una massa cistica, rialzata, iperriflettente che sporge dagli strati interni della retina. L’angiografia OCT (OCT-A) mostra un flusso sanguigno interno scarso, confermando le caratteristiche di una malformazione vascolare a basso flusso.
Il meccanismo esatto dell’emangioma cavernoso retinico non è completamente chiarito. Si ritiene che sia dovuto a un’anomalia dello sviluppo vascolare embrionale, ovvero un cambiamento amartomatoso localizzato del sistema venoso. Non sono stati identificati fattori di rischio ambientali per i casi sporadici.
Nei casi familiari (ereditarietà autosomica dominante), esiste un background genetico comune con le malformazioni cavernose cerebrali (CCM). Sono stati identificati tre geni causali per la CCM ed è stata riportata la loro associazione con malformazioni vascolari sistemiche, comprese le lesioni oculari1).
| Gene | Alias | Proteina codificata | Funzione principale |
|---|---|---|---|
| CCM1 | KRIT1 | Krev interaction trapped 1 | Adesione cellulare e omeostasi endoteliale vascolare |
| CCM2 | MGC4607 | Malcavernina | Legame con CCM1 e trasduzione del segnale |
| CCM3 | PDCD10 | Programmed cell death 10 | Controllo dell’apoptosi e della permeabilità vascolare |
Queste mutazioni genetiche sono coinvolte nell’adesione cellulare, nella trasduzione del segnale e nel controllo della permeabilità delle cellule endoteliali vascolari, portando a dilatazione venosa e fragilità della parete vascolare2).
I dati precisi sull’incidenza sono limitati, ma l’emangioma cavernoso retinico è considerato una malattia rara3). La prevalenza dell’emangioma cavernoso cerebrale nella popolazione generale è dello 0,1-0,5%, ma la proporzione con lesioni oculari è ancora minore. Ci sono pochi dati chiari sull’età di esordio e sul sesso. I casi familiari sono rari, ma sono state riportate diverse famiglie con associazione di emangiomi cerebrali, cutanei ed epatici.
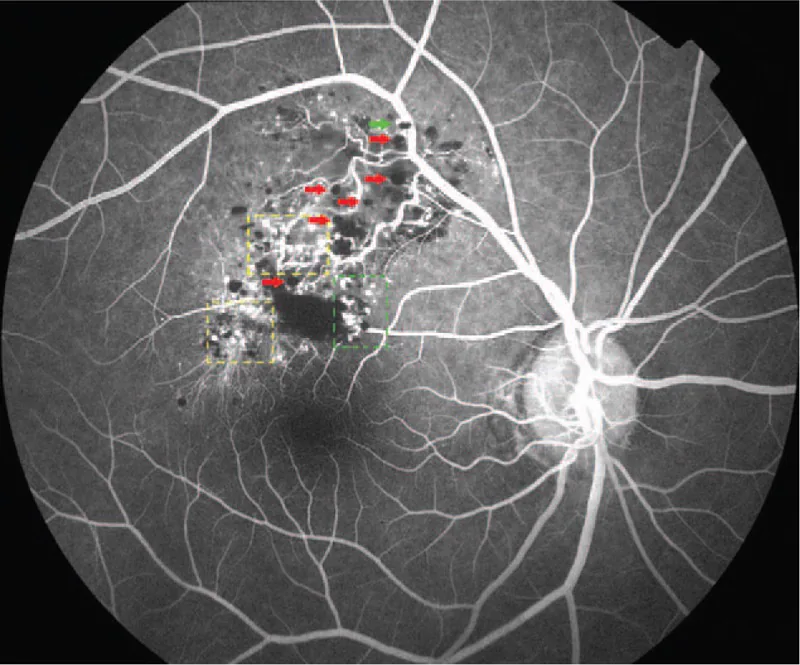
La diagnosi si basa principalmente sui reperti caratteristici del fundus (tumore multiloculare rosso scuro a grappolo d’uva) e sui reperti dell’angiografia retinica con fluoresceina (segno del cappuccio fluorescente, pattern di riempimento venoso con scarsa perdita di fluoresceina). Nei casi tipici non è necessaria una biopsia ed è possibile una diagnosi clinica.
Nell’angioma cavernoso retinico familiare (ereditarietà autosomica dominante) esiste un’associazione genetica con l’angioma cavernoso cerebrale (CCM). Le mutazioni dei geni CCM1, CCM2 e CCM3 sono una causa comune, portando a malformazioni vascolari multiple nella retina, cervello, pelle e fegato. Nei casi familiari, la RMN cerebrale con contrasto è obbligatoria per escludere lesioni cerebrali non scoperte (che possono causare epilessia o emorragia cerebrale). Nei casi sporadici, non è stata dimostrata alcuna associazione con lesioni cerebrali.
La diagnosi differenziale più importante dell’angioma cavernoso retinico è l’emangioblastoma retinico (malattia di von Hippel-Lindau). I reperti dell’angiografia retinica con fluoresceina sono il punto chiave per distinguere le due malattie.
| Malattia | Reperti del fondo oculare | Reperti FA | Complicanze sistemiche | Trattamento |
|---|---|---|---|---|
| Angioma cavernoso retinico | Massa multicistica rosso scuro (a grappolo d’uva) | Riempimento venoso lento, segno del cappuccio fluorescente, scarsa perdita | CCM (familiare) | Di solito osservazione |
| Emangioma capillare retinico (malattia di VHL) | Tumore semitrasparente arancione-rosso + vasi afferenti ed efferenti dilatati | Riempimento rapido in fase arteriosa, abbondante perdita di fluoresceina | Tumore VHL (carcinoma renale, ecc.) | Laser, chirurgia, anti-VEGF |
| Malattia di Coats | Distacco di retina essudativo, aneurismi capillari | Abbondante perdita dagli aneurismi capillari | Nessuna (più comune nei bambini) | Laser, chirurgia |
| Tumore proliferativo vascolare retinico | Massa periferica giallo-biancastra | Riempimento e perdita graduali | Nessuna | Laser, PDT |
Entrambe le malattie sono lesioni vascolari retiniche, ma possono essere chiaramente distinte con l’angiografia con fluoresceina. L’emangioma capillare retinico (malattia di VHL) è caratterizzato da un tumore arancione-rosso con vasi afferenti ed efferenti dilatati; all’FA si osserva un rapido riempimento nella fase arteriosa con abbondante perdita di fluoresceina. L’emangioma cavernoso retinico, invece, è caratterizzato da un lento riempimento nella fase venosa e dal segno del cappuccio fluorescente, con scarsa perdita di fluoresceina anche nelle fasi tardive. All’esame del fondo oculare, l’emangioma cavernoso retinico appare come una massa multiloculare rosso scuro a grappolo d’uva, e la differenziazione è solitamente possibile.
L’emangioma cavernoso retinico è non progressivo e di solito non richiede trattamento. Sulla lesione può formarsi una membrana fibroproliferativa, che può causare essudazione o emorragia da trazione. Nei pazienti asintomatici senza complicanze, si raccomanda un follow-up regolare con esame del fondo oculare.
Il trattamento viene preso in considerazione in caso di comparsa delle seguenti complicanze.
Vitrectomia: In caso di distacco di retina da trazione causato da membrana fibroproliferativa, o di emorragia vitreale associata a trazione, si può prendere in considerazione la vitrectomia. Poiché si tratta di una malformazione vascolare a basso flusso, il rischio di emorragia intra- e post-operatoria è considerato simile a quello della chirurgia retinica standard. La maggior parte dei dati sui risultati chirurgici proviene da report di casi, e le evidenze su larga scala sono scarse4).
Fotocoagulazione laser: Trattandosi di una malformazione vascolare a basso flusso, la risposta alla fotocoagulazione è scarsa. Al momento, non è attivamente raccomandata.
Nei casi familiari, oltre alla gestione delle lesioni oculari, è importante la gestione neurologica per l’angioma cavernoso cerebrale. Per le lesioni cerebrali asintomatiche, l’osservazione è la regola, ma in caso di crisi epilettiche o sintomi neurologici, si considera la terapia farmacologica o l’intervento chirurgico5).
Di solito non è necessario alcun trattamento. Si tratta di una malformazione vascolare non progressiva; nei casi asintomatici, la regola è il monitoraggio con esame del fondo oculare regolare. Solo in caso di complicanze da trazione dovute a membrana fibroproliferativa (distacco di retina da trazione, emorragia vitreale) è indicata la vitrectomia. La fotocoagulazione laser è poco efficace a causa del basso flusso e generalmente non viene eseguita. Nei casi familiari, oltre al follow-up oftalmologico, è necessaria una gestione sistemica degli angiomi cerebrali.
L’emangioma cavernoso retinico non è un tumore ma una malformazione vascolare. È un ammasso di vasi composto da cellule endoteliali, cellule muscolari lisce e cellule stromali, e non una proliferazione monoclonale (proliferazione tumorale). Nella classificazione rivista dell’ISSVA (International Society for the Study of Vascular Anomalies) del 2018, è classificato come malformazione venosa a basso flusso, distinta dalle malformazioni arterovenose (alto flusso)6).
Nei casi familiari (ereditarietà autosomica dominante), le mutazioni con perdita di funzione dei geni CCM svolgono un ruolo centrale. CCM1 (KRIT1) regola la via delle integrine coinvolta nell’adesione cellula-cellula, CCM2 (malcavernina) funge da proteina scaffold per CCM1, e CCM3 (PDCD10) è coinvolto nella regolazione dell’apoptosi e della permeabilità vascolare2). Queste mutazioni portano alla rottura dell’adesione cellula-cellula delle cellule endoteliali, alla transizione endotelio-mesenchimale, alla dilatazione vascolare e all’aumento della permeabilità.
Nei casi sporadici (non ereditari), si suggerisce un’anomalia locale dello sviluppo vascolare dovuta a mutazione somatica. Si ritiene che un’anomalia durante lo sviluppo vascolare retinico embrionale formi dilatazioni venose multicistiche a grappolo d’uva, ma i meccanismi dettagliati rimangono in gran parte sconosciuti.
Il fatto che sia una malformazione venosa a basso flusso determina le caratteristiche cliniche di questa malattia. A causa del basso flusso sanguigno, si verificano le seguenti caratteristiche:
Sulla superficie (lato vitreale) della malformazione vascolare può formarsi un cappuccio gliotico (gliotic cap) o una membrana fibroproliferativa. La contrazione di questa membrana può causare distacco di retina tractionale o emorragia vitreale. La membrana fibroproliferativa non è neoplastica, ma una proliferazione reattiva secondaria.
La comprensione dei meccanismi molecolari dovuti alle mutazioni del gene CCM sta progredendo. Negli studi animali, gli inibitori della segnalazione Rho/ROCK e gli inibitori della via PI3K/Akt/mTOR hanno mostrato risultati promettenti in modelli di angioma cavernoso cerebrale7). Sono necessarie ulteriori ricerche per l’applicazione alle lesioni oculari.
I test multigenici tramite sequenziamento di nuova generazione (NGS) si sono diffusi e vengono utilizzati per la diagnosi definitiva dei casi familiari, la diagnosi dei portatori e la consulenza genetica familiare. Anche l’interpretazione delle varianti patogene delle mutazioni del gene CCM si sta accumulando1).
Per quanto riguarda la gestione delle complicanze trazionali mediante vitrectomia, la letteratura a livello di case report e serie di casi si sta accumulando4,8). Non esistono studi randomizzati controllati su larga scala e sono auspicabili futuri studi prospettici.
Fischer A, Zalvide J, Faurobert E, Albiges-Rizo C, Tournier-Lasserve E.. Cerebral cavernous malformations: from CCM genes to endothelial cell homeostasis. Trends Mol Med. 2013;19(5):302-308. doi:10.1016/j.molmed.2013.02.004. PMID:23506982.
Couteulx SL, Jung HH, Labauge P, et al. Truncating mutations in CCM1, encoding KRIT1, cause hereditary cavernous angiomas. Nat Genet. 1999;23(2):189-193. doi:10.1038/13815.
Gass JDM. Cavernous hemangioma of the retina. A neuro-oculocutaneous syndrome. Am J Ophthalmol. 1971;71(4):799-814.
Messmer E, Font RL, Laqua H, Höpping W, Naumann GO. Cavernous hemangioma of the retina. Immunohistochemical and ultrastructural observations. Archives of ophthalmology (Chicago, Ill. : 1960). 1984;102(3):413-8. doi:10.1001/archopht.1984.01040030331031. PMID:6538410.
Haller JA Jr, Dortz J, Goldberg MF. Familial retinal cavernous hemangiomas. Arch Ophthalmol. 1979;97(5):879-883.
ISSVA Classification of Vascular Anomalies. International Society for the Study of Vascular Anomalies. 2018 update. Available at: https://www.issva.org.
Lewis RA, Cohen BH, Wise GN. Cavernous haemangioma of the retina and optic disc. A report of three cases and a review of the literature. Br J Ophthalmol. 1975;59(8):422-434.
Shields JA, Shields CL, Timmers E, et al. Spectrum of vitreoretinal surgery. Retina. 1992;12(1):1-11.