Sporadik (en sık)
Kalıtım şekli: Kalıtsal olmayan (sporadik)
Lezyon dağılımı: Tek taraflı, periferik retina
Klinik seyir: Genellikle ilerleyici değil, asemptomatik
Sistemik komplikasyonlar: Yok

Retinal kavernöz hemanjiyom (retinal cavernous hemangioma), düşük akımlı genişlemiş venlerin birikmesiyle oluşan retinal bir vasküler malformasyondur. Genellikle kalıtsal olmayan, tek taraflı, ilerleyici olmayan ve periferik retinada sporadik olarak ortaya çıkar. Optik disk veya makulada lezyon olan vakalar ile otozomal dominant kalıtım gösteren ve deri, santral sinir sistemi, karaciğer hemanjiyomlarının eşlik ettiği vakalar da bildirilmiştir.
Bu hastalık kesin olarak tümör değil, vasküler malformasyon olarak sınıflandırılır. Vasküler endotel hücreleri, düz kas hücreleri ve stromal hücrelerden oluşan bir damar kümesidir; tek bir hücrenin proliferasyonu değildir. Uluslararası Vasküler Anomali Çalışma Derneği (ISSVA) sınıflamasında düşük akımlı venöz malformasyon (venous malformation) olarak yer alır ve hemanjiyom-vasküler malformasyon tanı ve tedavi kılavuzlarında da yer alan bir hastalıktır.
Sporadik (en sık)
Kalıtım şekli: Kalıtsal olmayan (sporadik)
Lezyon dağılımı: Tek taraflı, periferik retina
Klinik seyir: Genellikle ilerleyici değil, asemptomatik
Sistemik komplikasyonlar: Yok
Ailesel (otozomal dominant kalıtım)
Kalıtım şekli: Otozomal dominant
Lezyon dağılımı: Tek veya iki göz, çoklu
Klinik seyir: Beyin, cilt, karaciğer komplikasyonları var
Sistemik komplikasyonlar: Serebral kavernöz malformasyon (CCM), cilt hemanjiomu, karaciğer hemanjiomu
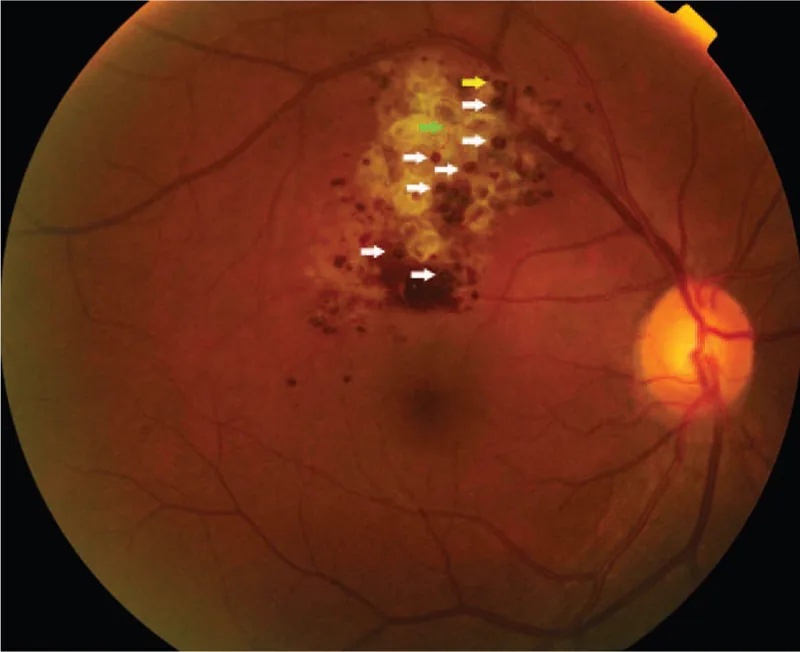
Lezyon periferle sınırlı olduğunda genellikle asemptomatiktir ve sıklıkla fundus muayenesinde tesadüfen saptanır. Lezyon optik disk veya makulayı etkilediğinde görme bozukluğu ve görme alanı defekti oluşabilir. Tümör üzerinde fibroproliferatif membran oluşursa ve traksiyon nedeniyle eksüdasyon veya kanama meydana gelirse görsel fonksiyon üzerindeki etki belirginleşir.
Fundus bulguları bu hastalığın tanısında en önemlisidir. Karakteristik bulgular aşağıda gösterilmiştir.
Floresein anjiyografi ayırıcı tanıda en yararlı incelemedir. Karakteristik bulgular aşağıda gösterilmiştir.
Optik koherens tomografide (OCT), retina iç katmanlarından çıkıntı yapan kistik, kabarık, yüksek yansıtıcı bir kitle olarak görülür. OCT anjiyografide (OCT-A) ise iç kan akımı zayıftır ve düşük akımlı vasküler malformasyon özellikleri doğrulanır.
Retinal kavernöz hemanjiomun kesin oluşum mekanizması tam olarak aydınlatılamamıştır. Embriyonik dönemde vasküler gelişim anomalisi, yani venöz sistemin lokal hamartomatöz değişikliğinin neden olduğu düşünülmektedir. Sporadik vakalar için çevresel risk faktörleri şu anda tanımlanmamıştır.
Ailesel vakalarda (otozomal dominant kalıtım), serebral kavernöz malformasyon (CCM) ile ortak bir genetik zemin bulunur. Serebral kavernöz malformasyonun nedensel genleri olarak aşağıdaki üç gen tanımlanmış olup, oküler lezyonları da içeren sistemik vasküler malformasyonlarla ilişkileri bildirilmiştir1).
| Gen | Diğer Adı | Kodlanan Protein | Ana İşlev |
|---|---|---|---|
| CCM1 | KRIT1 | Krev interaction trapped 1 | Hücre adezyonu ve vasküler endotel homeostazı |
| CCM2 | MGC4607 | Malcavernin | CCM1’e bağlanma ve sinyal iletimi |
| CCM3 | PDCD10 | Programmed cell death 10 | Apoptotik ve vasküler geçirgenlik kontrolü |
Bu gen mutasyonları, vasküler endotel hücrelerinin hücre adezyonu, sinyal iletimi ve geçirgenlik kontrolünde rol oynar ve venöz dilatasyon ile damar duvarında zayıflamaya yol açar2).
Kesin insidans verileri sınırlı olmakla birlikte, retinal kavernöz hemanjiyom nadir bir hastalık olarak kabul edilir3). Serebral kavernöz hemanjiyomun popülasyon bazlı prevalansı %0.1-0.5 olarak tahmin edilmektedir, ancak oküler tutulumu olan oran daha da düşüktür. Sık görülen yaş ve cinsiyet farkına dair net veri yoktur. Ailesel vakalar nadirdir, ancak beyin, deri ve karaciğer hemanjiyomları ile birlikte görülen birkaç aile rapor edilmiştir.
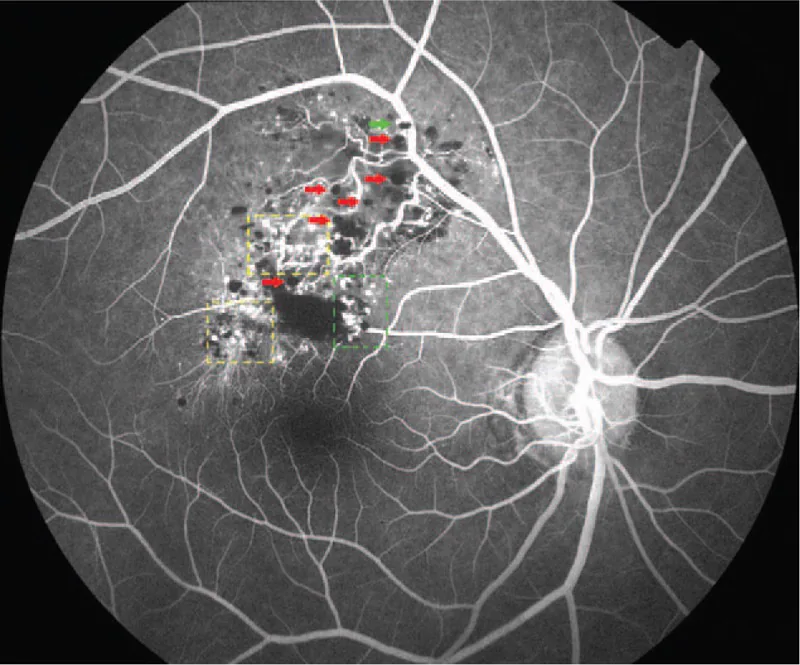
Tanı esas olarak karakteristik fundus bulguları (üzüm salkımı benzeri çok kistik koyu kırmızı kitle) ve floresein anjiyografi bulgularına (fluorescent cap sign, az floresan sızıntılı venöz dolum paterni) dayanır. Tipik vakalarda biyopsi gerekmez ve klinik tanı mümkündür.
Ailesel (otozomal dominant) retinal kavernöz anjiyomda, serebral kavernöz anjiyom (CCM) ile genetik ilişki vardır. CCM1, CCM2 ve CCM3 gen mutasyonları ortak nedendir ve retina, beyin, deri ve karaciğerde multipl vasküler malformasyonlara yol açar. Ailesel olgularda beyin kontrastlı MRG zorunludur; keşfedilmemiş beyin lezyonlarını (epilepsi veya beyin kanamasına neden olabilir) dışlamak önemlidir. Sporadik olgularda beyin lezyonlarıyla ilişki gösterilmemiştir.
Retinal kavernöz anjiyomun en önemli ayırıcı tanısı retinal kapiller hemanjiyomdur (VHL hastalığı). Floresein anjiyografi bulguları iki hastalığı ayırt etmede en önemli noktadır.
| Hastalık | Fundus Bulguları | FA Bulguları | Sistemik Komplikasyonlar | Tedavi |
|---|---|---|---|---|
| Retinal Kavernöz Anjiyom | Multikistik koyu kırmızı kitle (üzüm salkımı benzeri) | Yavaş venöz dolum, floresan cap işareti, az sızıntı | CCM (ailesel) | Genellikle takip |
| Retinal kapiller hemanjiyom (VHL hastalığı) | Turuncu-kırmızı yarı saydam tümör + genişlemiş giren ve çıkan damarlar | Arteriyel fazda hızlı dolum, yoğun floresan sızıntısı | VHL tümörleri (renal hücreli karsinom vb.) | Lazer, cerrahi, anti-VEGF |
| Coats hastalığı | Eksüdatif retina dekolmanı, kapiller anevrizma | Kapiller anevrizmadan yoğun sızıntı | Yok (çoğunlukla çocuklar) | Lazer, cerrahi |
| Retinal vasküler proliferatif tümör | Periferik sarı-beyaz kitle | Kademeli dolum ve sızıntı | Yok | Lazer, PDT |
Her iki hastalık da retinal vasküler lezyonlardır, ancak floresein anjiyografi bulguları ile net bir şekilde ayırt edilebilir. Retinal kapiller hemanjiyomda (VHL hastalığı), genişlemiş afferent ve efferent damarlarla birlikte turuncu-kırmızı tümör karakteristiktir ve FA’da arteriyel fazdan itibaren hızla dolar ve yoğun floresein sızıntısı görülür. Buna karşılık, retinal kavernöz hemanjiyom, venöz dolum fazında yavaş dolum ve floresan cap işareti ile karakterizedir ve geç dönemde bile floresein sızıntısı azdır. Fundus muayenesinde retinal kavernöz hemanjiyom, üzüm salkımı benzeri multiloküle koyu kırmızı kitle olarak görülür ve ayırım genellikle mümkündür.
Retinal kavernöz hemanjiyom ilerleyici değildir ve genellikle tedavi gerektirmez. Lezyon üzerinde fibrovasküler membran oluşabilir ve traksiyona bağlı eksüdasyon veya kanamaya neden olabilir. Asemptomatik ve komplikasyonsuz olgularda düzenli fundus muayenesi ile takip yapılır.
Aşağıdaki komplikasyonlar geliştiğinde tedavi düşünülür.
Vitrektomi: Fibrovasküler membrana bağlı traksiyonel retina dekolmanı veya traksiyona eşlik eden vitreus kanaması durumunda vitrektomi düşünülür. Düşük akımlı bir vasküler malformasyon olduğu için intraoperatif ve postoperatif kanama riski genel retina cerrahisi ile benzerdir. Cerrahi sonuçlar çoğunlukla olgu sunumları düzeyindedir ve büyük ölçekli kanıtlar sınırlıdır4).
Lazer fotokoagülasyon: Düşük akımlı vasküler malformasyon olması nedeniyle fotokoagülasyona yanıt zayıftır. Şu anda aktif olarak önerilmemektedir.
Ailesel vakalarda, göz lezyonlarının yönetiminin yanı sıra serebral kavernöz anjiyom için nörolojik yönetim de önemlidir. Beyin lezyonları için, asemptomatik ise takip temeldir, ancak epileptik nöbet veya nörolojik semptomlar ortaya çıkarsa ilaç tedavisi veya cerrahi müdahale düşünülür5).
Genellikle tedavi gerektirmez. Progresif olmayan bir vasküler malformasyondur ve asemptomatik vakalarda düzenli fundus muayenesi ile takip temel yaklaşımdır. Sadece fibroproliferatif membranın traksiyonel komplikasyonları (traksiyonel retina dekolmanı, vitreus hemorajisi) geliştiğinde vitrektomi cerrahisi endikedir. Lazer fotokoagülasyon düşük akım nedeniyle etkili değildir ve genellikle yapılmaz. Ailesel vakalarda oftalmolojik takibe ek olarak serebral anjiyomun sistemik yönetimi gereklidir.
Retinal kavernöz anjiyom bir tümör değil, vasküler malformasyondur. Vasküler endotel hücreleri, düz kas hücreleri ve interstisyel hücrelerden oluşan bir vasküler kitle olup, tek bir hücrenin proliferasyonu (tümöral büyüme) olmaması temel özelliğidir. ISSVA (Uluslararası Vasküler Anomali Çalışmaları Derneği) 2018 revizyon sınıflamasında düşük akımlı venöz malformasyon (venous malformation) olarak sınıflandırılır ve arteriyovenöz malformasyondan (yüksek akım) ayrılır6).
Ailesel (otozomal dominant) vakalarda, CCM genlerindeki fonksiyon kaybı mutasyonları merkezi rol oynar. CCM1 (KRIT1), hücreler arası adezyonda rol oynayan integrin yolunu düzenler. CCM2 (malcavernin), CCM1 için bir iskele proteini olarak işlev görür. CCM3 (PDCD10), apoptoz ve vasküler geçirgenliğin düzenlenmesinde rol oynar2). Bu mutasyonlar, vasküler endotel hücrelerinde hücreler arası adezyonun bozulmasına, endotelyal-mezenkimal geçişe (endothelial-to-mesenchymal transition), vazodilatasyona ve geçirgenlik artışına yol açar.
Sporadik (kalıtsal olmayan) vakalarda, somatik mutasyonların (somatic mutation) lokal vasküler gelişim anormalliğine neden olduğu düşünülmektedir. Embriyonik dönemde retinal vaskülogenez sırasındaki bir anormalliğin, üzüm salkımı benzeri multikistik venöz dilatasyonlar oluşturduğu varsayılmaktadır, ancak ayrıntılı mekanizma büyük ölçüde bilinmemektedir.
Düşük akımlı venöz vasküler malformasyon olması, bu hastalığın klinik özelliklerini belirler. Düşük kan akımı nedeniyle aşağıdaki özellikler ortaya çıkar:
Vasküler malformasyonun yüzeyinde (vitreus tarafında) gliotik cap veya fibröz proliferatif membran oluşabilir. Bu membranın kasılması traksiyonel retina dekolmanına veya vitreus kanamasına neden olabilir. Fibröz proliferatif membran neoplastik değildir, sekonder reaktif proliferasyondur.
CCM gen mutasyonlarının neden olduğu moleküler mekanizmaların aydınlatılması ilerlemektedir. Hayvan deneylerinde Rho/ROCK sinyal yolu inhibitörleri ve PI3K/Akt/mTOR yolu inhibitörleri serebral kavernöz anjiyom modellerinde umut verici sonuçlar göstermiştir7). Oküler lezyonlara uygulanması için daha fazla araştırmaya ihtiyaç vardır.
Yeni nesil dizileme (NGS) ile çoklu gen panel testi yaygınlaşmış ve ailesel vakaların kesin tanısı, taşıyıcı tanısı ve ailelere genetik danışmanlık verilmesinde kullanılmaktadır. CCM gen mutasyonlarının patojenik varyantlarının yorumlanması da birikmektedir1).
Traksiyonel komplikasyonların vitrektomi ile yönetimi konusunda vaka raporları ve vaka serileri düzeyinde literatür birikmektedir4,8). Büyük ölçekli randomize kontrollü çalışma bulunmamaktadır ve gelecekte prospektif çalışmalar beklenmektedir.
Fischer A, Zalvide J, Faurobert E, Albiges-Rizo C, Tournier-Lasserve E.. Cerebral cavernous malformations: from CCM genes to endothelial cell homeostasis. Trends Mol Med. 2013;19(5):302-308. doi:10.1016/j.molmed.2013.02.004. PMID:23506982.
Couteulx SL, Jung HH, Labauge P, et al. Truncating mutations in CCM1, encoding KRIT1, cause hereditary cavernous angiomas. Nat Genet. 1999;23(2):189-193. doi:10.1038/13815.
Gass JDM. Cavernous hemangioma of the retina. A neuro-oculocutaneous syndrome. Am J Ophthalmol. 1971;71(4):799-814.
Messmer E, Font RL, Laqua H, Höpping W, Naumann GO. Cavernous hemangioma of the retina. Immunohistochemical and ultrastructural observations. Archives of ophthalmology (Chicago, Ill. : 1960). 1984;102(3):413-8. doi:10.1001/archopht.1984.01040030331031. PMID:6538410.
Haller JA Jr, Dortz J, Goldberg MF. Familial retinal cavernous hemangiomas. Arch Ophthalmol. 1979;97(5):879-883.
ISSVA Classification of Vascular Anomalies. International Society for the Study of Vascular Anomalies. 2018 update. Available at: https://www.issva.org.
Lewis RA, Cohen BH, Wise GN. Cavernous haemangioma of the retina and optic disc. A report of three cases and a review of the literature. Br J Ophthalmol. 1975;59(8):422-434.
Shields JA, Shields CL, Timmers E, et al. Spectrum of vitreoretinal surgery. Retina. 1992;12(1):1-11.