Retinal astrositik hamartom, retinadaki astrositlerin aşırı çoğalması sonucu oluşan benign bir tümördür. Hamartom, o bölgede normalde bulunan olgun doku bileşenlerinin anormal oranlarda karışmasıyla oluşan tümöral lezyondur. Malignleşmez.
Bu hastalık, tüberoskleroz kompleksi (TSC) ile birlikte veya TSC olmaksızın sporadik olarak görülebilir. Tüberoskleroz, çeşitli organlarda hamartomlara neden olan otozomal dominant multisistemik bir hastalıktır; intrakraniyal lezyonlara bağlı epilepsi, ciltte adenoma sebaceum, renal anjiyomiyolipom ve retinal hamartom gibi çeşitli bulgularla ortaya çıkar.
Sorumlu genler TSC1 (kromozom 9) ve TSC2 (kromozom 16) olarak bilinir. TSC1 hamartin, TSC2 ise tüberin proteinini kodlar ve her ikisinin fonksiyon bozukluğu mTOR (mechanistic target of rapamycin) yolağının regülasyon bozukluğuna yol açar.
QTüberoskleroz olmadan da retinal astrositik hamartom oluşur mu?
A
TSC olmaksızın sporadik vakalar mevcuttur. Sporadik vakalarda bilateralite nadirdir ve sistemik komplikasyon olmadığı için oftalmolojik yönetim esas olur. Öte yandan, TSC’ye eşlik eden vakalarda multisistemik yönetim gerekir, bu nedenle kesin tanıda TSC ayırıcı tanısı önemlidir.
Qin X, Tao Y, Zhang Z. Retinal astrocytic hamartoma in tuberous sclerosis complex in an elderly person: a case report. BMC Ophthalmol. 2018;18(1):319. Figure 1. PMID: 30541513; PMCID: PMC6292060; DOI: 10.1186/s12886-018-0991-z. License: CC BY.
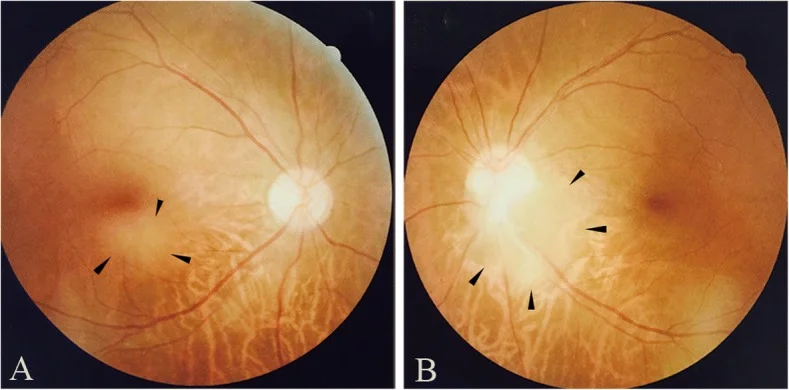
Sağ göz makula altı (a) ve sol göz optik disk temporal alt kenarında (b) retinal astrositomun fundus fotoğrafı. Bu bulgular, ‘2. Ana belirtiler ve klinik bulgular’ bölümünde ele alınan fundus bulgularına karşılık gelmektedir.
Retinal astrositik hamartom sıklıkla asemptomatiktir ve tüberoskleroz taraması sırasında fundus muayenesinde tesadüfen saptanır. Nadiren görme azalması veya uçuşan cisimler görülebilir. Tümör makulada yer alıyorsa görme üzerinde daha büyük etkisi olur.
Floresein anjiyografide (FAG), erken dönemde tümör içindeki ince damarlar görüntülenir. Geç dönemde floresein sızıntısı olmaması bu hastalığın karakteristik bulgusudur ve geç dönemde floresein sızıntısı gösteren retinoblastomadan önemli bir ayırıcı noktadır.
Optik koherens tomografide (OCT), kitle retina iç katmanlarından kabaran yüksek yansıtıcılı bir kitle olarak görülür. Kitlenin iç katman yapısının bozulması ve çevre normal retina ile sınırının nispeten belirgin olması karakteristiktir.
Tüberoz skleroz (TSC) otozomal dominant kalıtılır. Prevalansı yaklaşık 6.000-10.000’de 1’dir. TSC hastalarında retinal hamartom birlikteliği yaklaşık %50 olarak bildirilmiştir ve bilateral veya multipl olabilir.
TSC1 geni (9. kromozom q34) mutasyonları daha hafif seyirli iken, TSC2 geni (16. kromozom p13.3) mutasyonları daha ağır sistemik tutuluma yol açar. De novo mutasyonlar da sıktır ve aile öyküsü olmayan olgularda TSC dışlanamaz.
Sporadik (TSC dışı) olgularda genetik arka plan farklıdır ve sınırlı somatik mutasyonlar rol oynayabilir. Sporadik olgular genellikle tek taraflı ve tektir, sistemik komplikasyon eşlik etmez.
Multipl santral sinir sistemi, cilt veya böbrek lezyonları öyküsü
QCinsiyet veya yaşa bağlı bir yatkınlık var mı?
A
Belirgin bir cinsiyet farkı bildirilmemiştir. TSC’ye eşlik eden olgularda fundus lezyonları erken bebeklik döneminden itibaren görülebilir ve pediatrik taramanın bir parçası olarak fundus muayenesi önerilir. Sporadik olgular erişkinlerde de saptanabilir, ancak hepsi benign seyirlidir.
Retinal hamartom saptandığında, TSC tanı kriterlerine (Northrup revizyonu 2012 1)) göre sistemik değerlendirme yapılır. Majör kriterler arasında sebaceous adenom (yüz anjiyofibromu), epilepsi, serebral nodüller (Shagreen patch) bulunur. TSC’nin kesin tanısı için pediatri ve nöroloji ile iş birliği gereklidir.
Retinal astrositik hamartom genellikle büyümez; asemptomatik ve büyüme yoksa tedavi gereksizdir, takip esastır. Düzenli fundus muayenesi ile büyüme veya kanama varlığı kontrol edilir.
Göz lezyonu büyümüyorsa aktif tedavi gerekmez. Tüberoskleroz eşlik eden olgularda, santral sinir sistemi lezyonları (epilepsi, subependimal dev hücreli astrositom SEGA), renal anjiyomiyolipom gibi sistemik lezyonların yönetimi pediatri ve nöroloji uzmanlarıyla birlikte yapılmalıdır.
mTOR inhibitörü (everolimus), tüberoskleroza bağlı sistemik tümörlerde (SEGA ve renal anjiyomiyolipom) sigorta kapsamındadır ve sistemik TSC tedavisinin bir parçası olarak tümör küçültücü etkisi bildirilmiştir2)3). Retinal hamartom üzerinde doğrudan etkinlik kanıtı sınırlıdır, ancak sistemik TSC tedavisi bağlamında verilen olgularda küçülme bildirilmiştir.
QRetinal astrositik hamartom tedavisi gerekli midir?
A
Asemptomatik ve büyümeyen olgularda tedavi gerekmez, düzenli fundus muayenesi ile takip esastır. Yalnızca tekrarlayan kanama durumunda vitrektomi veya retina fotokoagülasyonu yapılır. Tüberoskleroz eşlik eden olgularda oftalmolojik yönetimle birlikte sistemik hastalık yönetimi önemlidir.
Retinal astrositik hamartomun patofizyolojisi, TSC1 (hamartin) ve TSC2 (tüberin) gen ürünlerinin işlev kaybından kaynaklanır.
Hamartin ve tüberin bir kompleks oluşturarak mTORC1 aktivitesini baskılayan tümör baskılayıcılar olarak işlev görür. TSC1 veya TSC2 mutasyonu bu mTOR düzenlemesinin kaybına ve mTORC1’in aşırı aktivasyonuna yol açar.
mTORC1’in aşırı aktivasyonu, S6 kinaz (S6K) ve 4E-BP1’in fosforilasyonu yoluyla hücre çoğalmasını, protein sentezini ve anjiyogenezi artırır. Sonuçta astrositler anormal şekilde çoğalır ve hamartom (iyi huylu kitle) oluşur.
Tümör oluşumunda “2-hit hipotezi”nin rol oynadığı düşünülmektedir. Germ hattında TSC1 veya TSC2’nin bir alelinde mutasyon taşıyan bireylerde, retina hücrelerinde somatik mutasyon (2. hit) meydana gelmesi, kalan normal alelin işlevini kaybetmesine (LOH: heterozigotluk kaybı) yol açar ve mTOR yolunun düzenlenmesi tamamen bozularak tümör oluşur.
Retinal astrositik hamartom, astrosit kaynaklı proliferasyondan oluşan iyi huylu bir kitledir ve malign dönüşüm göstermez. Histolojik olarak, anormal şekilde çoğalmış astrositler yoğun bir şekilde dizilir ve bazılarında kalsiyum birikimi (kalsifikasyon) eşlik eder. Hücre bölünme hızının düşük olması, tümörün yavaş büyümesi klinik özelliği ile uyumludur.
Everolimus, bir mTORC1 inhibitörü olarak tüberoskleroz ilişkili tümörlerin tedavisinde kullanılır. EXIST-1 çalışmasında (SEGA hedefli) 2), everolimus grubunda SEGA hacminde %50 veya daha fazla azalma oranı plasebo grubuna göre anlamlı derecede yüksekti. EXIST-2 çalışmasında (renal anjiyomiyolipom hedefli) 3) da benzer tümör küçülme etkisi rapor edilmiştir.
TSC ilişkili retinal hamartomda mTOR inhibitörlerinin oftalmik etkinliği konusunda şu anda yalnızca sınırlı vaka raporları bulunmaktadır. Sistemik TSC tedavisinin bir parçası olarak everolimus alan hastalarda retinal hamartomda küçülme gözlemlendiğine dair raporlar vardır, ancak büyük ölçekli prospektif çalışmalardan elde edilmiş kanıtlar henüz oluşturulmamıştır.
Fundus hamartomu genellikle büyümez, ancak nadiren büyüyerek kanama veya eksüdasyona neden olabilir. Büyüme risk faktörlerinin belirlenmesi, uzun dönemde görme değişikliklerinin tahmini ve uygun müdahale zamanının belirlenmesi için daha fazla vaka birikimi ve prospektif çalışmalara ihtiyaç vardır.
TSC olmaksızın görülen sporadik vakaların genetik arka planı ve moleküler mekanizması tam olarak aydınlatılamamıştır. Somatik mutasyonlara bağlı lokal mTOR yolu anormalliğinin rol oynayabileceği düşünülmektedir, ancak detaylı mekanizmanın aydınlatılması gelecekteki araştırma konusudur.
Rowley SA, O’Callaghan FJ, Osborne JP.. Ophthalmic manifestations of tuberous sclerosis: a population based study. Br J Ophthalmol. 2001;85(4):420-423. doi:10.1136/bjo.85.4.420. PMID:11264130; PMCID:PMC1723924.
Zimmer-Galler IE, Robertson DM.. Long-term observation of retinal lesions in tuberous sclerosis. Am J Ophthalmol. 1995;119(3):318-324. doi:10.1016/s0002-9394(14)71174-2. PMID:7872393.
Neil J Lucchese, Morton F Goldberg. Iris and Fundus Pigmentary Changes in Tuberous Sclerosis. J Pediatr Ophthalmol Strabismus. 1981;18(6):45-46. doi:10.3928/0191-3913-19811101-12.
Makale metnini kopyalayıp tercih ettiğiniz yapay zeka asistanına yapıştırabilirsiniz.
Makale panoya kopyalandı
Aşağıdaki yapay zeka asistanlarından birini açın ve kopyalanan metni sohbet kutusuna yapıştırın.