O hamartoma astrocítico retiniano (retinal astrocytic hamartoma) é um tumor benigno resultante da proliferação excessiva de astrócitos (células gliais estreladas) na retina. Hamartoma é uma lesão tumoral composta por componentes de tecido maduro normalmente presentes no local, mas em proporções anormais. Não se torna maligno.
Esta doença pode estar associada ao complexo de esclerose tuberosa (tuberous sclerosis complex; TSC) ou ocorrer esporadicamente sem TSC. O complexo de esclerose tuberosa é uma doença multissistêmica autossômica dominante que causa hamartomas em vários órgãos, manifestando-se com convulsões devido a lesões intracranianas, adenomas sebáceos na pele, angiomiolipomas renais, hamartomas retinianos, entre outros sintomas.
Dois genes causadores são conhecidos: TSC1 (cromossomo 9) e TSC2 (cromossomo 16). TSC1 codifica a hamartina, TSC2 codifica a tuberina, e a disfunção de ambos leva ao distúrbio de regulação da via mTOR (mechanistic target of rapamycin).
QO hamartoma astrocítico retiniano pode ocorrer sem esclerose tuberosa?
A
Sim, existem casos esporádicos sem TSC. Nos casos esporádicos, o envolvimento bilateral é raro e não há complicações sistêmicas, sendo o manejo oftalmológico o principal. Já nos casos associados ao TSC, é necessário o manejo multiorgânico, portanto, é importante diferenciar o TSC no diagnóstico.
Qin X, Tao Y, Zhang Z. Retinal astrocytic hamartoma in tuberous sclerosis complex in an elderly person: a case report. BMC Ophthalmol. 2018;18(1):319. Figure 1. PMID: 30541513; PMCID: PMC6292060; DOI: 10.1186/s12886-018-0991-z. License: CC BY.
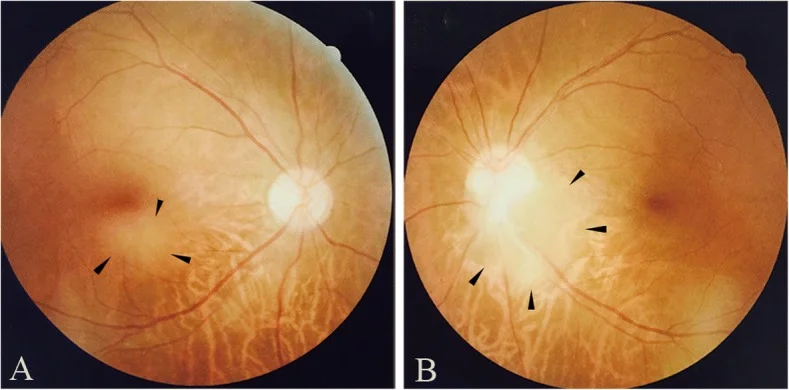
Fotografia de fundo de olho mostrando astrocitoma retiniano na região sub-macular do olho direito (a) e na borda inferotemporal do disco óptico do olho esquerdo (b). Corresponde aos achados de fundo discutidos na seção “2. Principais sintomas e achados clínicos”.
O astrocitoma retiniano é frequentemente assintomático, sendo descoberto incidentalmente durante exame de fundo de olho de triagem para esclerose tuberosa. Raramente ocorre diminuição da acuidade visual ou moscas volantes. Se o tumor estiver na mácula, o impacto na visão é significativo.
Na angiografia fluoresceínica (FAG), os microvasos dentro do tumor são visualizados na fase inicial. Na fase tardia, não se observa extravasamento de fluoresceína, o que é um achado característico desta doença e um importante ponto de diferenciação do retinoblastoma, que mostra extravasamento na fase tardia.
Na tomografia de coerência óptica (OCT), o tumor é visualizado como uma massa hiperrefletiva que se projeta das camadas internas da retina. Caracteriza-se por desorganização da estrutura interna do tumor e bordas relativamente nítidas em relação à retina normal circundante.
O complexo de esclerose tuberosa (TSC) é uma doença autossômica dominante. A prevalência é de aproximadamente 1 em 6.000 a 10.000 pessoas. A incidência de hamartoma retiniano em pacientes com TSC é relatada em cerca de 50%, podendo ser bilateral ou múltiplo.
Mutações no gene TSC1 (cromossomo 9q34) tendem a causar sintomas mais leves, enquanto mutações no TSC2 (cromossomo 16p13.3) estão associadas a lesões sistêmicas mais graves. Mutações de novo são comuns, e o TSC não pode ser descartado mesmo na ausência de história familiar.
Em casos esporádicos (não TSC), o histórico genético é diferente, e mutações somáticas localizadas podem estar envolvidas. Casos esporádicos são geralmente unilaterais e solitários, sem complicações sistêmicas.
História familiar ou diagnóstico confirmado de complexo de esclerose tuberosa (TSC)
Mutações genéticas conhecidas de TSC (TSC1 ou TSC2)
História de múltiplas lesões no sistema nervoso central, pele ou rins
QExiste tendência de ocorrência por sexo ou idade?
A
Não há diferença clara de sexo relatada. Em casos associados ao TSC, lesões de fundo de olho podem ser detectadas precocemente após o nascimento, e o exame de fundo de olho é recomendado como parte da triagem pediátrica. Casos esporádicos também podem ser encontrados em adultos, mas todos têm curso benigno.
A doença mais importante a ser diferenciada é o retinoblastoma. Em crianças com lesão retiniana elevada e branca, o retinoblastoma deve ser excluído primeiro.
Quando um hamartoma retiniano é descoberto, realiza-se uma avaliação sistêmica com base nos critérios diagnósticos de TSC (revisão de Northrup 2012 1)). Os critérios maiores incluem adenoma sebáceo (angiofibroma facial), epilepsia e nódulos cerebrais (mancha de Shagreen). A colaboração com pediatria e neurologia é essencial para o diagnóstico definitivo de TSC.
O hamartoma astrocítico retiniano geralmente não cresce; se assintomático e sem crescimento, não é necessário tratamento e a observação é a base. Exames de fundo de olho periódicos são realizados para verificar crescimento ou hemorragia.
Se a lesão ocular não aumentar, não é necessário tratamento ativo. Em casos associados à esclerose tuberosa, o manejo das lesões sistêmicas (como epilepsia, astrocitoma subependimal de células gigantes SEGA, angiomiolipoma renal) deve ser realizado em conjunto com pediatras e neurologistas.
Inibidores de mTOR (everolimo) têm cobertura de seguro para tumores sistêmicos associados à esclerose tuberosa (como SEGA e angiomiolipoma renal), e foi relatado efeito de redução tumoral como parte do tratamento sistêmico da TSC2)3). As evidências de eficácia direta em hamartomas retinianos são limitadas, mas há relatos de redução em casos administrados no contexto do tratamento sistêmico da TSC.
QO tratamento do astrocitoma hamartoma retiniano é necessário?
A
Se assintomático e sem crescimento, o tratamento não é necessário, e o acompanhamento periódico com exame de fundo de olho é a base. A vitrectomia ou fotocoagulação retiniana é realizada apenas em casos de sangramento recorrente. Em casos associados à esclerose tuberosa, o manejo da doença sistêmica é importante em paralelo ao manejo oftalmológico.
A patogênese do astrocitoma hamartoma retiniano decorre da perda de função dos produtos dos genes TSC1 (hamartina) e TSC2 (tuberina).
A hamartina e a tuberina formam um complexo que atua como supressor tumoral ao inibir a atividade do complexo mTOR1 (mTORC1). Mutações em TSC1 ou TSC2 levam à perda desse controle do mTOR, resultando em hiperativação do mTORC1.
A hiperativação do mTORC1, por meio da fosforilação da S6 quinase (S6K) e 4E-BP1, promove proliferação celular, síntese proteica e angiogênese. Como resultado, os astrócitos proliferam anormalmente formando um hamartoma (massa benigna).
Acredita-se que a “hipótese dos dois eventos” (2-hit hypothesis) esteja envolvida no desenvolvimento do tumor. Em indivíduos com uma mutação alélica em TSC1 ou TSC2 na linhagem germinativa, a ocorrência de uma mutação somática (segundo evento) nas células da retina leva à perda da função do alelo normal restante (LOH: perda de heterozigosidade), resultando na ruptura completa da regulação da via mTOR e na formação do tumor.
O hamartoma astrocítico da retina é uma massa benigna composta por proliferação derivada de astrócitos, e não sofre transformação maligna. Histologicamente, os astrócitos anormalmente proliferados estão densamente arranjados, com algumas áreas contendo depósitos de cálcio (calcificação). A baixa taxa de divisão celular corresponde à característica clínica de crescimento lento do tumor.
O everolimo é usado como inibidor de mTORC1 no tratamento de tumores associados à esclerose tuberosa. No estudo EXIST-1 (direcionado a SEGA) 2), a proporção de redução do volume de SEGA em 50% ou mais foi significativamente maior no grupo everolimo em comparação com o grupo placebo. Um efeito semelhante de redução tumoral foi relatado no estudo EXIST-2 (direcionado a angiomiolipoma renal) 3).
Quanto à eficácia oftalmológica dos inibidores de mTOR em hamartomas retinianos associados a TSC, atualmente existem apenas relatos de casos limitados. Há relatos de redução do hamartoma retiniano em pacientes que receberam everolimo como parte do tratamento sistêmico de TSC, mas evidências de grandes ensaios prospectivos não foram estabelecidas.
Prognóstico e desafios do acompanhamento de longo prazo
O hamartoma retiniano geralmente não aumenta, mas em casos raros pode aumentar e causar hemorragia ou exsudação. A identificação de fatores de risco para aumento, a previsão de mudanças na visão a longo prazo e a determinação do momento adequado para intervenção requerem maior acúmulo de casos e estudos prospectivos.
Patofisiologia do hamartoma astrocítico retiniano esporádico
O background genético e os mecanismos moleculares dos casos esporádicos sem TSC não foram totalmente elucidados. A possibilidade de envolvimento de anormalidades locais da via mTOR devido a mutações somáticas foi sugerida, mas a elucidação dos mecanismos detalhados é um tópico de pesquisa futura.
Rowley SA, O’Callaghan FJ, Osborne JP.. Ophthalmic manifestations of tuberous sclerosis: a population based study. Br J Ophthalmol. 2001;85(4):420-423. doi:10.1136/bjo.85.4.420. PMID:11264130; PMCID:PMC1723924.
Zimmer-Galler IE, Robertson DM.. Long-term observation of retinal lesions in tuberous sclerosis. Am J Ophthalmol. 1995;119(3):318-324. doi:10.1016/s0002-9394(14)71174-2. PMID:7872393.
Neil J Lucchese, Morton F Goldberg. Iris and Fundus Pigmentary Changes in Tuberous Sclerosis. J Pediatr Ophthalmol Strabismus. 1981;18(6):45-46. doi:10.3928/0191-3913-19811101-12.
Copie o texto do artigo e cole no assistente de IA de sua preferência.
Artigo copiado para a área de transferência
Abra um assistente de IA abaixo e cole o texto copiado na conversa.