แฮมาร์โทมาของเซลล์แอสโทรไซต์ในจอตา (retinal astrocytic hamartoma) เป็นเนื้องอกชนิดไม่ร้ายแรงที่เกิดจากการเพิ่มจำนวนของเซลล์แอสโทรไซต์ (astrocytes) ในจอตา
อาจเกิดร่วมกับโรคทูเบอรัส สเกลอโรซิส (TSC ) หรือเกิดแบบประปราย คาดว่าประมาณ 50% ของผู้ป่วย TSC มีภาวะนี้
ทำให้เกิดรอยโรคสีขาวนูนขึ้นในจอตา แบ่งเป็น 2 ชนิด: ชนิดผลหม่อน (mulberry) และชนิดราบ
การไม่มีการรั่วของฟลูออเรสซีน ในระยะปลายในการตรวจฟลูออเรสซีน แองจิโอกราฟีเป็นจุดแยกที่สำคัญจากรีติโนบลาสโตมา
โดยปกติไม่โตและไม่ต้องรักษา เฉพาะในกรณีที่มีเลือดออกซ้ำจึงทำการผ่าตัดวุ้นตา หรือจอตาเลเซอร์
ความผิดปกติในการควบคุมวิถี mTOR เนื่องจากการกลายพันธุ์ของยีน TSC 1/TSC 2 เป็นพื้นฐานของพยาธิสรีรวิทยา
การพยากรณ์โรคของรอยโรคทางตาดีโดยทั่วไป แต่การจัดการรอยโรคในระบบประสาทส่วนกลางและไตส่งผลต่อการรอดชีวิต
แฮมาร์โทมาของเซลล์แอสโทรไซต์ในจอตา (retinal astrocytic hamartoma) เป็นเนื้องอกชนิดไม่ร้ายแรงที่เกิดจากการเพิ่มจำนวนมากเกินไปของเซลล์แอสโทรไซต์ (astrocytes) ในจอตา แฮมาร์โทมา (hamartoma) คือรอยโรคคล้ายเนื้องอกที่ประกอบด้วยส่วนประกอบของเนื้อเยื่อเจริญเต็มที่ซึ่งปกติมีอยู่บริเวณนั้น แต่มีสัดส่วนผิดปกติ ไม่กลายเป็นมะเร็ง
โรคนี้สามารถเกิดร่วมกับโรคทูเบอรัส สเกลอโรซิส (tuberous sclerosis complex; TSC ) หรือเกิดแบบประปรายโดยไม่มี TSC โรคทูเบอรัส สเกลอโรซิสเป็นโรคหลายระบบที่ถ่ายทอดแบบออโตโซมัลเด่น ทำให้เกิดแฮมาร์โทมาในอวัยวะต่างๆ แสดงอาการด้วยอาการชักจากรอยโรคในกะโหลกศีรษะ ต่อมไขมันที่ผิวหนัง แองจิโอไมโอลิโพมาของไต แฮมาร์โทมาของจอตา และอาการอื่นๆ
ยีนก่อโรคที่รู้จักมีสองยีน: TSC 1 (บนโครโมโซม 9) และ TSC 2 (บนโครโมโซม 16) TSC 1 เข้ารหัสแฮมาร์ติน (hamartin) TSC 2 เข้ารหัสทูเบอริน (tuberin) และความผิดปกติของทั้งสองทำให้เกิดความผิดปกติในการควบคุมวิถี mTOR (mechanistic target of rapamycin)
Q
แฮมาร์โทมาของเซลล์แอสโทรไซต์ในจอตาสามารถเกิดขึ้นได้โดยไม่มีโรคทูเบอรัส สเกลอโรซิสหรือไม่?
A
ใช่ มีกรณีประปรายที่ไม่มี TSC ในกรณีประปราย การเกิดสองตานั้นพบได้น้อย และไม่มีภาวะแทรกซ้อนทางระบบ ดังนั้นการจัดการทางจักษุวิทยาจึงเป็นหลัก ในขณะที่กรณีที่เกิดร่วมกับ TSC จำเป็นต้องจัดการหลายอวัยวะ ดังนั้นการแยก TSC ในการวินิจฉัยจึงมีความสำคัญ
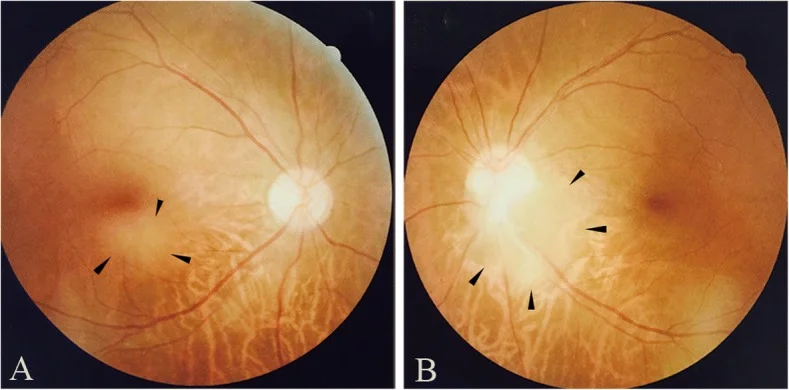
Qin X, Tao Y, Zhang Z. Retinal astrocytic hamartoma in tuberous sclerosis complex in an elderly person: a case report. BMC Ophthalmol. 2018;18(1):319. Figure 1. PMID: 30541513; PM
CI D: PMC6292060; DOI: 10.1186/s12886-018-0991-z. License: CC BY.
ภาพถ่ายจอตาที่แสดงแอสโตรไซโตมาจอตาในบริเวณใต้จุดรับภาพของตาขวา (a) และที่ขอบด้านล่างขมับของจานประสาทตา ของตาซ้าย (b) สอดคล้องกับผลการตรวจจอตา ที่กล่าวถึงในหัวข้อ “2. อาการหลักและผลการตรวจทางคลินิก”
แอสโตรไซโตมาจอตามักไม่มีอาการ ตรวจพบโดยบังเอิญระหว่างการตรวจคัดกรองจอตาสำหรับโรคทูเบอรัส สเกลอโรซิส ไม่ค่อยพบการมองเห็น ลดลงหรือจุดลอย หากเนื้องอกอยู่ที่จุดรับภาพ จะมีผลต่อการมองเห็น อย่างมาก
ทำให้เกิดรอยโรคสีขาวนูนลักษณะเฉพาะบนจอตา จำแนกตามลักษณะภายนอกเป็น 2 ชนิดดังนี้
ชนิดผลหม่อน (mulberry type)
ลักษณะภายนอก : ก้อนนูนรูปโดมที่มีพื้นผิวไม่เรียบ รูปร่างลักษณะเฉพาะคล้าย “ผลหม่อน”
การกลายเป็นปูน : มักมีการกลายเป็นปูนร่วมด้วย ความแตกต่างจากการกลายเป็นปูนของรีติโนบลาสโตมา คือมีสีเหลืองเข้มกว่า
ขอบ : สะท้อนการแบ่งตัวของเซลล์ที่ช้า ดังนั้นการนูนจึงเล็กน้อยและขอบลาดเอียง
ชนิดแบน
ลักษณะภายนอก : รอยโรคแบน สีขาวโปร่งแสง คล้ายรีติโนบลาสโตมา
ความสำคัญของการวินิจฉัยแยกโรค : เนื่องจากความคล้ายคลึงทางลักษณะภายนอกกับรีติโนบลาสโตมา การวินิจฉัยแยกโรคจึงต้องประเมินอย่างระมัดระวัง
การกลายเป็นปูน : มักไม่มีการกลายเป็นปูนเมื่อเทียบกับชนิดผลหม่อน
ในการตรวจฟลูออเรสซีน แองจิโอกราฟี (FAG ) จะเห็นหลอดเลือดขนาดเล็กภายในเนื้องอกในระยะแรก ในระยะหลังไม่พบการรั่วของฟลูออเรสซีน ซึ่งเป็นลักษณะเฉพาะของโรคนี้ และเป็นจุดแยกสำคัญจากรีติโนบลาสโตมา ที่แสดงการรั่วในระยะหลัง
ในการตรวจ optical coherence tomography (OCT ) เนื้องอกจะปรากฏเป็นก้อนสะท้อนแสงสูงที่ยื่นออกมาจากชั้นในของจอประสาทตา มีลักษณะเด่นคือโครงสร้างภายในของเนื้องอกไม่เป็นระเบียบ และขอบเขตค่อนข้างชัดเจนเมื่อเทียบกับจอประสาทตา ปกติโดยรอบ
โรคทูเบอรัส สเกลอโรซิส (TSC ) เป็นโรคที่ถ่ายทอดทางพันธุกรรมแบบ autosomal dominant ความชุกประมาณ 1 ใน 6,000–10,000 คน อุบัติการณ์ของ hamartoma จอประสาทตา ในผู้ป่วย TSC รายงานประมาณ 50% และอาจพบได้ทั้งสองข้างหรือหลายตำแหน่ง
การกลายพันธุ์ของยีน TSC 1 (โครโมโซม 9q34) มักทำให้เกิดอาการไม่รุนแรง ในขณะที่การกลายพันธุ์ของ TSC 2 (โครโมโซม 16p13.3) มักทำให้เกิดรอยโรคทั่วร่างกายที่รุนแรงกว่า การกลายพันธุ์ใหม่ (de novo mutation) ก็พบได้บ่อย ดังนั้นจึงไม่สามารถแยกโรค TSC ออกได้แม้ไม่มีประวัติครอบครัว
ในกรณีประปราย (ไม่ใช่ TSC ) ภูมิหลังทางพันธุกรรมจะแตกต่างกัน และอาจเกี่ยวข้องกับการกลายพันธุ์ของโซมาติกเฉพาะที่ กรณีประปรายมักเป็นข้างเดียวและเดี่ยว ไม่มีภาวะแทรกซ้อนทั่วร่างกาย
ประวัติครอบครัวหรือการวินิจฉัยที่แน่ชัดของโรคทูเบอรัส สเกลอโรซิส (TSC )
การกลายพันธุ์ของยีน TSC ที่ทราบ (TSC 1 หรือ TSC 2)
ประวัติรอยโรคหลายแห่งในระบบประสาทส่วนกลาง ผิวหนัง หรือไต
Q
มีความแตกต่างของแนวโน้มการเกิดโรคตามเพศหรืออายุหรือไม่?
A
ไม่มีความแตกต่างทางเพศที่ชัดเจนรายงาน ในกรณีที่เกี่ยวข้องกับ TSC สามารถตรวจพบรอยโรคที่จอประสาทตา ได้ตั้งแต่ระยะแรกหลังคลอด และแนะนำให้ตรวจอวัยวะตาเป็นส่วนหนึ่งของการตรวจคัดกรองในเด็ก กรณีประปรายอาจพบได้ในผู้ใหญ่เช่นกัน แต่ทั้งหมดมีแนวทางที่ไม่ร้ายแรง
การวินิจฉัย astrocytoma จอประสาทตา ทำได้โดยการรวมลักษณะเฉพาะของจอประสาทตา และลักษณะทางระบบของ tuberous sclerosis
จุดแยกโรคในการตรวจจอประสาทตา :
รอยโรคสีขาวนูนคล้ายผลหม่อนหรือแบบราบ
ไม่มีการรั่วของฟลูออเรสซีน ระยะปลายในการตรวจฟลูออเรสซีน แองจิโอกราฟี
การนูนเล็กน้อยและขอบเรียบ
ไม่มีการขยายใหญ่ขึ้นระหว่างการติดตามผล
สีเหลืองเข้มแม้มีการกลายเป็นปูน
โรคที่สำคัญที่สุดที่ต้องแยกคือ retinoblastoma ในเด็กที่มีรอยโรคจอประสาทตา สีขาวนูน ต้องแยก retinoblastoma ออกก่อน
ชื่อโรค จุดแยกโรค Retinoblastoma โตเร็ว มีการรั่วของฟลูออเรสซีน ระยะปลาย มีการกลายเป็นปูน (สีขาว) Retinocytoma ไม่มีการดำเนินโรค ลักษณะการหดตัวแบบกลายเป็นปูน ไม่โต หลอดเลือดคอรอยด์ เดี่ยว สีส้มแดง, เพิ่มความคมชัดในการตรวจหลอดเลือดด้วยฟลูออเรสซีน
เมื่อพบแฮมาร์โทมาจอประสาทตา ให้ประเมินร่างกายโดยรวมตามเกณฑ์การวินิจฉัย TSC (Northrup revision 2012 1) ) เกณฑ์หลัก ได้แก่ ต่อมไขมัน (angiofibroma ที่ใบหน้า), โรคลมชัก, และก้อนในสมอง (Shagreen patch) การร่วมมือกับกุมารแพทย์และอายุรแพทย์ระบบประสาทเป็นสิ่งจำเป็นสำหรับการวินิจฉัยที่แน่นอนของ TSC
แฮมาร์โทมาจอประสาทตา อาจเป็นอาการเริ่มแรกของโรคทูเบอรัส สเกลอโรซิส หากจักษุแพทย์ตรวจพบโรคนี้เป็นครั้งแรก แนะนำให้ตรวจร่างกายอย่างละเอียดรวมถึง MRI สมอง, อัลตราซาวนด์ช่องท้อง, และการประเมินผิวหนัง ในเด็ก การส่งต่อไปยังอายุรแพทย์ระบบประสาทหรือกุมารแพทย์เป็นสิ่งสำคัญ
แฮมาร์โทมาแอสโตรไซต์จอประสาทตา มักไม่โต; หากไม่มีอาการและไม่โต ไม่จำเป็นต้องรักษา การติดตามผลเป็นพื้นฐาน ตรวจอวัยวะภายในตาเป็นระยะเพื่อดูการโตหรือเลือดออก
ในกรณีที่มีเลือดออกซ้ำ ให้พิจารณาการรักษาต่อไปนี้
การผ่าตัดน้ำวุ้นตา
ข้อบ่งชี้ : เลือดออกซ้ำทำให้เกิดเลือดออกในน้ำวุ้นตา หรือมีการเปลี่ยนแปลงของจอประสาทตา จากการดึงรั้ง
วัตถุประสงค์ : กำจัดเลือดออกในน้ำวุ้นตา และปลดการดึงรั้งเพื่อรักษาการทำงานของการมองเห็น
การพยากรณ์โรค : ด้วยข้อบ่งชี้การผ่าตัดที่เหมาะสม สามารถคาดหวังการรักษาระดับการมองเห็น ได้
การจี้แสงเลเซอร์จอตา
ข้อบ่งชี้ : การรักษาเสริมในกรณีที่มีการรั่วซึมหรือเลือดออกเล็กน้อยจากขอบเนื้องอก
วัตถุประสงค์ : ปิดแหล่งเลือดออกจากหลอดเลือดเนื้องอกและป้องกันการตกเลือดซ้ำ
ข้อควรระวัง : ในรอยโรคที่จุดภาพชัด ควรพิจารณาข้อบ่งชี้ของการจี้แสงอย่างรอบคอบ
หากรอยโรคทางตาไม่ขยายใหญ่ขึ้น ไม่จำเป็นต้องรักษาเชิงรุก ในกรณีที่ร่วมกับโรคทูเบอรัส สเกลอโรซิส ควรจัดการรอยโรคทั่วร่างกาย (เช่น โรคลมชัก, อะสโตรไซโตมาใต้เยื่อบุโพรงสมองชนิดเซลล์ยักษ์ SEGA, แองจิโอไมโอลิโพมาของไต) ร่วมกับกุมารแพทย์และอายุรแพทย์ระบบประสาท
ยายับยั้ง mTOR (เอเวอโรลิมัส) มีสิทธิ์เบิกได้ในเนื้องอกทั่วร่างกายที่ร่วมกับโรคทูเบอรัส สเกลอโรซิส (เช่น SEGA และแองจิโอไมโอลิโพมาของไต) และมีรายงานผลการลดขนาดเนื้องอกซึ่งเป็นส่วนหนึ่งของการรักษา TSC ทั่วร่างกาย2) 3) หลักฐานประสิทธิผลโดยตรงต่อแฮมาร์โทมาจอตามีจำกัด แต่มีรายงานการลดขนาดในผู้ป่วยที่ได้รับยาในบริบทของการรักษา TSC ทั่วร่างกาย
Q
จำเป็นต้องรักษาแอสโตรไซโตมา แฮมาร์โทมาจอตาหรือไม่?
A
หากไม่มีอาการและไม่ขยายใหญ่ขึ้น ไม่จำเป็นต้องรักษา การติดตามผลเป็นระยะด้วยการตรวจอวัยวะรับภาพเป็นพื้นฐาน การผาตัดน้ำวุ้นตา หรือการจี้แสงเลเซอร์จอตาจะทำเฉพาะในกรณีที่มีเลือดออกซ้ำ ในกรณีที่ร่วมกับโรคทูเบอรัส สเกลอโรซิส การจัดการโรคทั่วร่างกายมีความสำคัญควบคู่ไปกับการจัดการทางจักษุวิทยา
พยาธิกำเนิดของแอสโตรไซโตมา แฮมาร์โทมาจอตาเกิดจากการสูญเสียการทำงานของผลิตภัณฑ์ยีน TSC 1 (ฮามาร์ติน) และ TSC 2 (ทูเบอริน)
ฮามาร์ตินและทูเบอรินสร้างสารเชิงซ้อนซึ่งทำหน้าที่เป็นยีนต้านเนื้องอกโดยยับยั้งการทำงานของสารเชิงซ้อน mTOR1 (mTORC1) การกลายพันธุ์ของ TSC 1 หรือ TSC 2 ทำให้สูญเสียการควบคุม mTOR นี้ ส่งผลให้ mTORC1 ถูกกระตุ้นมากเกินไป
การกระตุ้น mTORC1 มากเกินไป ผ่านฟอสโฟรีเลชันของ S6 ไคเนส (S6K) และ 4E-BP1 ส่งเสริมการเพิ่มจำนวนเซลล์ การสังเคราะห์โปรตีน และการสร้างหลอดเลือดใหม่ ผลที่ตามมาคือแอสโตรไซต์เพิ่มจำนวนผิดปกติจนเกิดเป็นแฮมาร์โทมา (ก้อนเนื้อไม่ร้าย)
เชื่อกันว่า “สมมติฐาน 2-hit” (2-hit hypothesis) เกี่ยวข้องกับการเกิดเนื้องอก ในบุคคลที่มีการกลายพันธุ์ของอัลลีลหนึ่งใน TSC 1 หรือ TSC 2 ในสายพันธุ์สืบพันธุ์ การเกิดการกลายพันธุ์ของเซลล์ร่างกาย (hit ที่สอง) ในเซลล์จอประสาทตา ทำให้สูญเสียการทำงานของอัลลีลปกติที่เหลืออยู่ (LOH: loss of heterozygosity) ส่งผลให้การควบคุมวิถี mTOR พังทลายลงอย่างสมบูรณ์และเกิดเนื้องอกขึ้น
แฮมาร์โทมาเซลล์แอสโทรไซต์จอประสาทตา เป็นก้อนเนื้อไม่ร้ายแรงที่ประกอบด้วยการเจริญเติบโตที่มาจากเซลล์แอสโทรไซต์ และไม่เปลี่ยนเป็นมะเร็ง ทางจุลกายวิภาค เซลล์แอสโทรไซต์ที่เจริญเติบโตผิดปกติจะเรียงตัวกันอย่างหนาแน่น โดยบางบริเวณมีแคลเซียมสะสม (การกลายเป็นปูน) อัตราการแบ่งเซลล์ที่ช้าสอดคล้องกับลักษณะทางคลินิกที่เนื้องอกโตช้า
เนื้อหาต่อไปนี้อยู่ในขั้นตอนการวิจัยหรือการทดลองทางคลินิกในปัจจุบัน และไม่ใช่การรักษามาตรฐานที่สามารถรับได้ในโรงพยาบาลทั่วไป เป็นข้อมูลอ้างอิงสำหรับผู้เชี่ยวชาญเกี่ยวกับความก้าวหน้าทางการแพทย์ในอนาคต
เอเวอโรลิมัส (everolimus) ถูกใช้เป็นยายับยั้ง mTORC1 ในการรักษาเนื้องอกที่เกี่ยวข้องกับโรคทูเบอรัส สเกลอโรซิส ในการทดลอง EXIST-1 (ซึ่งศึกษา SEGA) 2) สัดส่วนของการลดปริมาตร SEGA ลง 50% หรือมากกว่านั้นสูงกว่าอย่างมีนัยสำคัญในกลุ่มที่ได้รับเอเวอโรลิมัสเมื่อเทียบกับกลุ่มที่ได้รับยาหลอก รายงานผลการลดขนาดเนื้องอกที่คล้ายกันยังพบในการทดลอง EXIST-2 (ซึ่งศึกษา angiomyolipoma ของไต) 3)
เกี่ยวกับประสิทธิภาพทางจักษุวิทยาของยายับยั้ง mTOR ต่อแฮมาร์โทมาจอประสาทตา ที่เกี่ยวข้องกับ TSC ในปัจจุบันมีเพียงรายงานผู้ป่วยจำนวนจำกัดเท่านั้น มีรายงานการหดตัวของแฮมาร์โทมาจอประสาทตา ในผู้ป่วยที่ได้รับเอเวอโรลิมัสเป็นส่วนหนึ่งของการรักษา TSC ทั่วร่างกาย แต่ยังไม่มีหลักฐานจากการทดลองไปข้างหน้าขนาดใหญ่
โดยทั่วไปแฮมาร์โทมาจอประสาทตา ไม่ขยายใหญ่ขึ้น แต่ในบางกรณีที่พบไม่บ่อยอาจขยายใหญ่ขึ้นและทำให้เกิดเลือดออกหรือสารคัดหลั่ง การระบุปัจจัยเสี่ยงต่อการขยายใหญ่ การทำนายการเปลี่ยนแปลงของการมองเห็น ในระยะยาว และการตัดสินใจช่วงเวลาที่เหมาะสมในการแทรกแซง จำเป็นต้องมีการสะสมผู้ป่วยและการศึกษาไปข้างหน้าเพิ่มเติม
พื้นฐานทางพันธุกรรมและกลไกระดับโมเลกุลของผู้ป่วยชนิดประปรายที่ไม่มี TSC ยังไม่ได้รับการอธิบายอย่างเพียงพอ มีการเสนอว่าความผิดปกติเฉพาะที่ของวิถี mTOR เนื่องจากการกลายพันธุ์ของเซลล์ร่างกายอาจเกี่ยวข้อง แต่การอธิบายกลไกโดยละเอียดเป็นหัวข้อของการวิจัยในอนาคต
Northrup H, Krueger DA; International Tuberous Sclerosis Complex Consensus Group. Tuberous sclerosis complex diagnostic criteria update: recommendations of the 2012 International Tuberous Sclerosis Complex Consensus Conference. Pediatr Neurol. 2013;49(4):243-254.
Franz DN, Belousova E, Sparagana S, Bebin EM, Frost M, Kuperman R, Witt O, Kohrman MH, Flamini JR, Wu JY, Curatolo P, de Vries PJ, Whittemore VH, Thiele EA, Ford JP, Shah G, Cauwel H, Lebwohl D, Sahmoud T, Jozwiak S.. Efficacy and safety of everolimus for subependymal giant cell astrocytomas associated with tuberous sclerosis complex (EXIST-1): a multicentre, randomised, placebo-controlled phase 3 trial. Lancet. 2013;381(9861):125-132. doi:10.1016/s0140-6736(12)61134-9. PMID:23158522.
Bissler JJ, Kingswood JC, Radzikowska E, Zonnenberg BA, Frost M, Belousova E, Sauter M, Nonomura N, Brakemeier S, de Vries PJ, Whittemore VH, Chen D, Sahmoud T, Shah G, Lincy J, Lebwohl D, Budde K.. Everolimus for angiomyolipoma associated with tuberous sclerosis complex or sporadic lymphangioleiomyomatosis (EXIST-2): a multicentre, randomised, double-blind, placebo-controlled trial. Lancet. 2013;381(9869):817-824. doi:10.1016/s0140-6736(12)61767-x. PMID:23312829.
Shields CL, Shields JA, Kiratli H, et al. Familial retinal astrocytoma. Retina. 2002;22(1):95-97.
Rowley SA, O’Callaghan FJ, Osborne JP.. Ophthalmic manifestations of tuberous sclerosis: a population based study. Br J Ophthalmol. 2001;85(4):420-423. doi:10.1136/bjo.85.4.420. PMID:11264130; PMCI D:PMC1723924.
Zimmer-Galler IE, Robertson DM.. Long-term observation of retinal lesions in tuberous sclerosis. Am J Ophthalmol. 1995;119(3):318-324. doi:10.1016/s0002-9394(14)71174-2. PMID:7872393.
Neil J Lucchese, Morton F Goldberg. Iris and Fundus Pigmentary Changes in Tuberous Sclerosis. J Pediatr Ophthalmol Strabismus. 1981;18(6):45-46. doi:10.3928/0191-3913-19811101-12.