El hamartoma astrocítico retiniano es un tumor benigno causado por la proliferación excesiva de astrocitos retinianos. Un hamartoma es una lesión tumoral compuesta por componentes de tejido maduro normalmente presentes en ese sitio, pero en proporciones anormales. No se maligniza.
Esta enfermedad puede asociarse con el complejo de esclerosis tuberosa (TSC) o presentarse de forma esporádica sin TSC. La esclerosis tuberosa es un trastorno multisistémico autosómico dominante que causa hamartomas en varios órganos, manifestándose con síntomas diversos como epilepsia debida a lesiones intracraneales, angiofibromas faciales, angiomiolipomas renales y hamartomas retinianos.
Se conocen dos genes causales: TSC1 (cromosoma 9) y TSC2 (cromosoma 16). TSC1 codifica la hamartina y TSC2 la tuberina, y la disfunción de ambos conduce a la desregulación de la vía mTOR (diana mecanicista de la rapamicina).
Q¿Puede ocurrir un hamartoma astrocítico retiniano sin esclerosis tuberosa?
A
Existen casos esporádicos sin TSC. En los casos esporádicos, la bilateralidad es rara y, al no haber complicaciones sistémicas, el manejo es principalmente oftalmológico. En cambio, en los casos asociados a TSC se requiere manejo multiorgánico, por lo que es importante diferenciar la TSC para el diagnóstico definitivo.
Qin X, Tao Y, Zhang Z. Retinal astrocytic hamartoma in tuberous sclerosis complex in an elderly person: a case report. BMC Ophthalmol. 2018;18(1):319. Figure 1. PMID: 30541513; PMCID: PMC6292060; DOI: 10.1186/s12886-018-0991-z. License: CC BY.
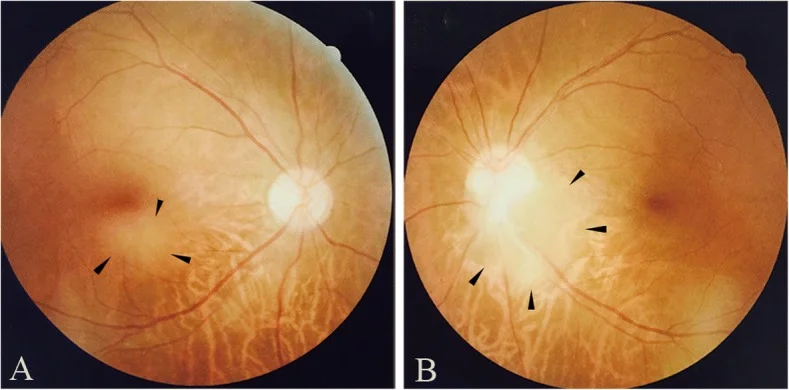
Fotografías de fondo de ojo de astrocitoma retiniano observado en la región subfoveal del ojo derecho (a) y en el borde inferotemporal del disco óptico del ojo izquierdo (b). Corresponden a los hallazgos de fondo de ojo discutidos en la sección “2. Síntomas principales y hallazgos clínicos”.
El hamartoma astrocítico retiniano a menudo es asintomático y se descubre incidentalmente durante el examen de fondo de ojo de detección para esclerosis tuberosa. En raras ocasiones, puede ocurrir disminución de la agudeza visual o moscas volantes. Cuando el tumor se encuentra en la mácula, el impacto en la visión es mayor.
Aparecen lesiones elevadas blancas características en el fondo de ojo. Según las diferencias en la apariencia, se clasifican en los siguientes dos tipos.
Tipo mora (mulberry)
Apariencia: Elevación en forma de cúpula con superficie irregular. Morfología característica que recuerda a una mora.
Calcificación: A menudo acompañada de calcificación. Una característica distintiva de la calcificación del retinoblastoma es un tono amarillento más fuerte.
Borde: Reflejando una división celular lenta, la elevación es leve y el borde asciende suavemente.
Tipo plano
Apariencia: Lesión plana, blanca y translúcida, que se asemeja al retinoblastoma en apariencia.
Importancia de la diferenciación: Debido a su similitud en apariencia con el retinoblastoma, se requiere una evaluación cuidadosa para el diagnóstico diferencial.
Calcificación: A menudo no se acompaña de calcificación en comparación con el tipo mora.
En la angiografía fluoresceínica (FAG), se visualizan finos vasos intratumorales en la fase temprana. La ausencia de fuga en fase tardía es un hallazgo característico de esta enfermedad y un punto importante de diferenciación del retinoblastoma, que muestra fuga en fase tardía.
En la tomografía de coherencia óptica (OCT), el tumor aparece como una masa hiperreflectante que sobresale de las capas internas de la retina. Las características incluyen la alteración de la estructura laminar interna y un borde relativamente claro con la retina normal circundante.
El complejo de esclerosis tuberosa (TSC) sigue un patrón de herencia autosómico dominante. La prevalencia se estima en aproximadamente 1 de cada 6,000 a 10,000 personas. La tasa reportada de hamartoma retiniano en pacientes con TSC es de alrededor del 50%, y puede ser bilateral y multifocal.
Las mutaciones en el gen TSC1 (cromosoma 9q34) tienden a causar síntomas más leves, mientras que las mutaciones en el gen TSC2 (cromosoma 16p13.3) son más propensas a provocar afectación sistémica grave. Las mutaciones de novo también son comunes, por lo que no se puede descartar TSC incluso sin antecedentes familiares.
En los casos esporádicos (no TSC), el trasfondo genético es diferente y pueden estar implicadas mutaciones somáticas localizadas. Los casos esporádicos suelen ser unilaterales y únicos, sin complicaciones sistémicas.
Antecedentes familiares o diagnóstico confirmado de complejo de esclerosis tuberosa (TSC)
Mutaciones genéticas conocidas en TSC (TSC1 o TSC2)
Antecedentes de múltiples lesiones del sistema nervioso central, cutáneas o renales
Q¿Existe una tendencia de aparición según el sexo o la edad?
A
No se ha reportado una diferencia clara de sexo. En los casos asociados a TSC, las lesiones del fondo de ojo pueden detectarse temprano después del nacimiento, y se recomienda el examen de fondo de ojo como parte del cribado pediátrico. Los casos esporádicos pueden descubrirse incluso en adultos, pero todos siguen un curso benigno.
El diagnóstico del hamartoma astrocítico retiniano se realiza combinando los hallazgos característicos del fondo de ojo con los hallazgos sistémicos de la esclerosis tuberosa.
Puntos clave para el diagnóstico diferencial en el examen de fondo de ojo:
Lesiones elevadas blancas con aspecto de mora o planas
La enfermedad diferencial más importante es el retinoblastoma. Cuando se encuentra una lesión retiniana elevada blanca en un niño, primero se debe descartar el retinoblastoma.
Cuando se encuentra un hamartoma retiniano, se debe realizar una evaluación sistémica basada en los criterios diagnósticos de TSC (revisión de Northrup 20121)). Los criterios mayores incluyen angiofibroma facial, epilepsia y parche de shagreen. La colaboración con pediatría y neurología es esencial para el diagnóstico definitivo de TSC.
El hamartoma astrocítico retiniano generalmente no aumenta de tamaño. Si es asintomático y sin crecimiento, no se requiere tratamiento y la observación es la norma. Se deben realizar exámenes fundoscópicos regulares para verificar si hay crecimiento o hemorragia.
Indicaciones de tratamiento y procedimientos quirúrgicos
Si la lesión ocular no aumenta de tamaño, no es necesario un tratamiento agresivo. En casos asociados con esclerosis tuberosa, el manejo de lesiones sistémicas como lesiones del sistema nervioso central (epilepsia, astrocitoma subependimario de células gigantes SEGA) y angiomiolipoma renal debe realizarse junto con pediatras y neurólogos.
Inhibidores de mTOR (everolimus) están cubiertos por el seguro para tumores sistémicos asociados con esclerosis tuberosa (SEGA y angiomiolipoma renal), y se han reportado efectos de reducción tumoral como parte del tratamiento sistémico de TSC2)3). La evidencia de eficacia directa contra hamartomas retinianos es limitada, pero hay informes de reducción en casos administrados en el contexto del tratamiento sistémico de TSC.
Q¿Es necesario tratar el hamartoma astrocítico retiniano?
A
Si es asintomático y no aumenta de tamaño, no es necesario tratamiento, y la base es la observación periódica mediante examen de fondo de ojo. Solo se realiza vitrectomía o fotocoagulación retiniana cuando hay sangrado recurrente. En casos asociados con esclerosis tuberosa, el manejo de la enfermedad sistémica es importante en paralelo con el manejo oftalmológico.
La patogenia del hamartoma astrocítico retiniano se debe a la pérdida de función de los productos génicos TSC1 (hamartina) y TSC2 (tuberina).
La hamartina y la tuberina forman un complejo y funcionan como supresores tumorales que inhiben la actividad del complejo 1 de mTOR (mTORC1). Cuando esta función reguladora de mTOR se pierde debido a mutaciones en TSC1 o TSC2, mTORC1 se hiperactiva.
La hiperactivación de mTORC1 promueve la proliferación celular, la síntesis de proteínas y la angiogénesis a través de la fosforilación de la quinasa S6 (S6K) y 4E-BP1. Como resultado, los astrocitos proliferan anormalmente, formando hamartomas (tumores benignos).
Se cree que la “hipótesis de dos impactos” está involucrada en el desarrollo tumoral. En individuos con una mutación germinal en un alelo de TSC1 o TSC2, una mutación somática (segundo impacto) en las células de la retina provoca la pérdida de función del alelo normal restante (LOH: pérdida de heterocigosidad), lo que resulta en una desregulación completa de la vía mTOR y la formación del tumor.
El hamartoma astrocítico retiniano es un tumor benigno compuesto por proliferación de astrocitos y no sufre transformación maligna. Histológicamente, los astrocitos anormalmente proliferados están densamente dispuestos, con algunos depósitos de calcio (calcificación). La baja tasa de división celular corresponde a la característica clínica de que el tumor solo crece lentamente.
7. Investigación más reciente y perspectivas futuras
Everolimus, un inhibidor de mTORC1, se utiliza para tratar tumores asociados al complejo de esclerosis tuberosa. En el ensayo EXIST-1 (dirigido a SEGA) 2), la proporción de pacientes con una reducción ≥50% en el volumen de SEGA fue significativamente mayor en el grupo de everolimus en comparación con el grupo placebo. El ensayo EXIST-2 (dirigido a angiomiolipoma renal) 3) también informó efectos similares de reducción tumoral.
En cuanto a la eficacia oftálmica de los inhibidores de mTOR para los hamartomas retinianos asociados a TSC, actualmente solo existen informes de casos limitados. Se ha informado de reducción de hamartomas retinianos en pacientes que recibieron everolimus como parte del tratamiento sistémico de TSC, pero no se ha establecido evidencia de ensayos prospectivos a gran escala.
Pronóstico y desafíos en el seguimiento a largo plazo
Los hamartomas del fondo de ojo generalmente no aumentan de tamaño, pero en raras ocasiones pueden crecer y causar hemorragia o exudación. La identificación de factores de riesgo de crecimiento, la predicción de cambios en la visión a largo plazo y la determinación del momento adecuado para la intervención requieren una mayor acumulación de casos y estudios prospectivos.
Patología del hamartoma astrocítico retiniano esporádico
El trasfondo genético y los mecanismos moleculares de los casos esporádicos sin TSC no se comprenden completamente. Se ha sugerido que las anomalías locales de la vía mTOR debidas a mutaciones somáticas pueden estar involucradas, pero la elucidación de los mecanismos detallados es un tema de investigación futura.
Rowley SA, O’Callaghan FJ, Osborne JP.. Ophthalmic manifestations of tuberous sclerosis: a population based study. Br J Ophthalmol. 2001;85(4):420-423. doi:10.1136/bjo.85.4.420. PMID:11264130; PMCID:PMC1723924.
Zimmer-Galler IE, Robertson DM.. Long-term observation of retinal lesions in tuberous sclerosis. Am J Ophthalmol. 1995;119(3):318-324. doi:10.1016/s0002-9394(14)71174-2. PMID:7872393.
Neil J Lucchese, Morton F Goldberg. Iris and Fundus Pigmentary Changes in Tuberous Sclerosis. J Pediatr Ophthalmol Strabismus. 1981;18(6):45-46. doi:10.3928/0191-3913-19811101-12.
Copia el texto del artículo y pégalo en el asistente de IA que prefieras.
Artículo copiado al portapapeles
Abre un asistente de IA abajo y pega el texto copiado en el chat.