El retinoblastoma es un tumor maligno de la retina en lactantes y niños pequeños. Resulta de la transformación maligna y proliferación de células retinianas inmaduras, y se considera una enfermedad monogénica causada por mutaciones en el gen RB1 ubicado en el brazo largo del cromosoma 13 (13q14.2). No hay predilección por sexo, y el 95% se diagnostica antes de los 5 años.
La incidencia es de 1 en 15,000 a 23,000 nacidos vivos, con 70-80 nuevos casos por año en Japón. La proporción de casos unilaterales a bilaterales es de 3:2, y los casos bilaterales se diagnostican más temprano (media de 8 meses) que los unilaterales (media de 21 meses). En países desarrollados, la tasa de supervivencia a 5 años para la enfermedad intraocular es superior al 95%.
El retinoblastoma se clasifica ampliamente en dos tipos según el tipo de mutación genética.
Clasificación
Tipo de mutación
Características de la enfermedad
Riesgo genético
Hereditario (mutación germinal)
Mutación germinal de RB1
Frecuentemente tumores bilaterales y multifocales
50% de riesgo de herencia en los hijos
No hereditario (mutación somática)
Mutación somática en una célula de la retina
Tumor unilateral y solitario
Sin herencia a la siguiente generación
Hereditario (mutación germinal): La primera mutación está presente en todas las células del cuerpo. El cáncer se desarrolla cuando ocurre una segunda mutación. Tiende a causar tumores bilaterales y multifocales, y se hereda en 1 de cada 2 hijos (50%). Existe riesgo de cánceres secundarios como osteosarcoma (15.7% a los 20 años).
No hereditario (mutación somática): Cuando ocurren mutaciones en ambos alelos del gen RB1 en una sola célula de la retina. Se presenta como un tumor unilateral y solitario, sin riesgo de herencia a la siguiente generación.
Sin embargo, algunos casos unilaterales incluyen mutaciones germinales de RB1. No se debe negar la herencia solo por ser unilateral; es necesario interpretar los antecedentes familiares, la edad de inicio y el número de tumores basándose en el asesoramiento genético y la evaluación genética. 1)
La estadificación está directamente relacionada con la estrategia del tratamiento de preservación ocular.
Estadio
Estado de la lesión
Tasa estimada de preservación ocular
T1 (lesión intraocular temprana)
Confinado al ojo, sin progresión
>90%
T2 (lesión intraocular avanzada)
Progresión intraocular
Aproximadamente 50%
T3
Lesión avanzada con invasión extraocular
Aproximadamente 10%
Q¿El retinoblastoma es hereditario?
A
Alrededor del 40% de los casos son hereditarios (mutación germinal de RB1), con un 50% de probabilidad de transmitirse a los hijos. El 60% restante son no hereditarios (solo mutación somática) sin riesgo de herencia para la siguiente generación. Los casos hereditarios tienden a ser bilaterales y multifocales. Se recomienda realizar pruebas genéticas y consejería genética en el momento del diagnóstico.
Aerts I, et al. Retinoblastoma. Orphanet J Rare Dis. 2006 Aug 25; 1:31. Figure 2. PMCID: PMC1586012. License: CC BY.
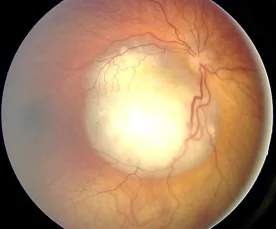
Imagen de fondo de ojo de retinoblastoma observado como una lesión elevada blanca con abundante vascularización, que muestra hallazgos típicos que causan leucocoria. Corresponde a los hallazgos de fondo de ojo y leucocoria tratados en la sección “2. Síntomas principales y hallazgos clínicos”.
En muchos casos, el tumor crece dentro del ojo y se descubre por leucocoria. Cuando ocurre en la mácula, puede causar mala visión y estrabismo, lo que lleva a su detección. En niños mayores, la disminución subjetiva de la visión puede ser el síntoma inicial, mientras que en niños pequeños, frotarse el ojo con mala visión puede ser el primer signo.
Síntomas tempranos
Leucocoria: El síntoma inicial más frecuente. El tumor se agranda dentro del ojo y la pupila se ve blanca.
Estrabismo: Causado por mala visión debido a un tumor en la mácula. El ojo con mala visión se desvía hacia afuera.
Disminución subjetiva de la visión: Se observa en niños mayores.
Frotarse el ojo: Se observa en niños pequeños con mala visión.
Síntomas avanzados
Opacidad corneal y aumento de la presión intraocular: Causados por desplazamiento del cristalino debido al tumor o aumento de la presión intraocular por glaucoma neovascular (NVG).
Inyección conjuntival e hinchazón palpebral: Se observan como signos inflamatorios.
Dolor: Aparece con el aumento de la presión intraocular o la necrosis tumoral.
Proptosis: Se observa cuando ocurre extensión extraocular.
Se observa una lesión elevada, blanca y vascularizada; si hay calcificación, el diagnóstico es sencillo. A menudo se acompaña de siembra vítrea (células tumorales que se desprenden y se diseminan al vítreo).
La prueba del reflejo rojo es fundamental para el cribado de enfermedades oculares en lactantes. La evaluación es normal si ambas pupilas son del mismo tamaño y muestran un reflejo amarillo-naranja brillante y simétrico. Si el reflejo es oscuro o demasiado brillante, o si hay asimetría entre los ojos, se considera anormal y requiere evaluación adicional.
Q¿Una pupila blanca siempre significa retinoblastoma?
A
Las causas de leucocoria (pupila blanca) incluyen no solo retinoblastoma, sino también vasculatura fetal persistente (hiperplasia vítrea primaria persistente), retinopatía del prematuro, enfermedad de Coats y muchas otras. Sin embargo, si se observa leucocoria, la prioridad máxima es una evaluación oftalmológica rápida para descartar retinoblastoma. El retraso en el diagnóstico afecta directamente el pronóstico; por lo tanto, si se sospecha, se recomienda la derivación el mismo día.
La causa son mutaciones en el gen RB1 en el brazo largo del cromosoma 13 (13q14.2). El gen RB1 produce la proteína RB1 (proteína del retinoblastoma), que desempeña un papel clave en la regulación de la división celular.
Cada célula tiene dos copias del gen; una mutación en una sola copia no afecta la función, pero cuando ambas copias están mutadas, la célula pierde el control de la división y se vuelve maligna (teoría de los dos impactos, hipótesis de Knudson).
Hereditario (mutación germinal): El primer impacto es una mutación en la línea germinal (presente en todas las células). Un segundo impacto en una célula somática conduce al cáncer. Son comunes los tumores bilaterales y multifocales.
No hereditario (mutación somática): Tanto el primer como el segundo impacto ocurren en una sola célula somática. Esto da lugar a tumores unilaterales y unifocales.
Los antecedentes familiares son el factor de riesgo más importante. Las definiciones de riesgo recomendadas por la AAOOP (Asociación Estadounidense de Oncología y Patología Oftálmica) se muestran a continuación 1).
Categoría de riesgo
Definición
Valor de riesgo
Alto
Padre con Rb bilateral, o familiar de primer o segundo grado portador de mutación germinal en RB1
En casos hereditarios, se debe prestar atención al riesgo de segundos cánceres. El osteosarcoma es un ejemplo típico, que ocurre en el 15.7% de los casos hereditarios en 20 años. Los segundos cánceres a menudo se desarrollan después de la adolescencia.
Rumboldt Z, Dodig D, Galluzzi P, et al. Retinoblastoma and beyond: pediatric orbital mass lesions. Neuroradiology. 2025;67(2):469-492. Figure 2. PMID: 39729290; PMCID: PMC11893699; DOI: 10.1007/s00234-024-03517-6. License: CC BY.
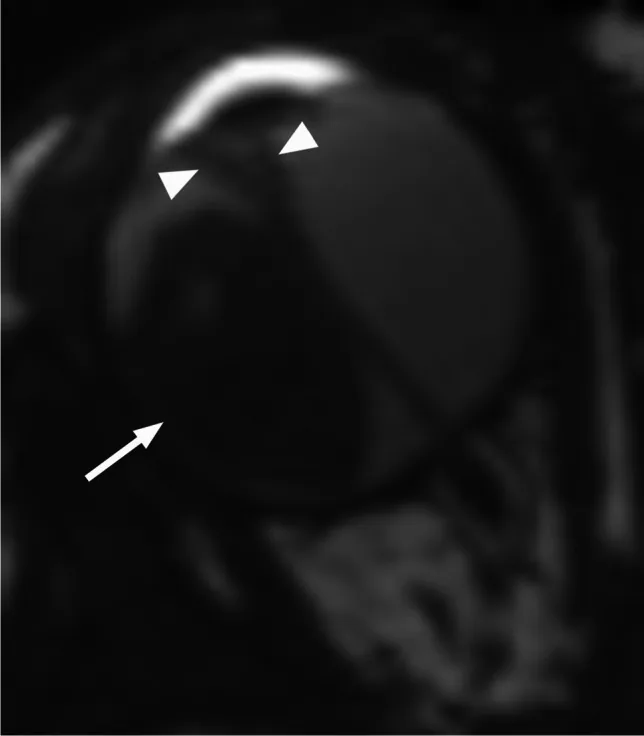
La RM axial potenciada en T2 muestra un retinoblastoma exofítico (flecha) con desprendimiento de retina secundario en forma de V (punta de flecha). Esto corresponde a los patrones de crecimiento exofítico y endofítico y a los hallazgos de RM discutidos en la sección “4. Diagnóstico y métodos de exploración.”
No se realiza biopsia de tumores intraoculares. Las lesiones intraoculares pueden observarse directamente a través de tejidos transparentes, y el diagnóstico clínico es muy preciso. Además, la biopsia de tumores intraoculares puede causar diseminación extraocular de células tumorales, con un riesgo inevitable de metástasis. Cuando se planifica un tratamiento de conservación ocular, el tratamiento se inicia basándose en el diagnóstico clínico.
Examen de fondo de ojo (principal): lesión elevada blanquecina vascularizada con calcificación para diagnóstico clínico definitivo
Ecografía: confirmación de tumor sólido y calcificación. Tenga en cuenta que la calcificación es menos común en niños mayores de 5 años.
RM: señal intermedia en T1, señal ligeramente baja en T2, con realce. Esencial para evaluar la invasión del nervio óptico y la extensión extraocular.
RM craneal: el cribado es obligatorio porque aproximadamente el 3% de los casos bilaterales desarrollan retinoblastoma trilateral (tumor de la glándula pineal).
TC: excelente para detectar calcificaciones pero implica exposición a radiación. Si se dispone de RM, la TC es complementaria.
Retinopatía del prematuro: Antecedentes de parto prematuro y bajo peso al nacer ayudan al diagnóstico diferencial
Enfermedad de Coats: Exudado subretiniano amarillento; patrón vascular tumoral diferente
Hamartoma astrocítico: Diferenciado por presencia de vasos tumorales, localización en OCT y crecimiento
Cisticercosis intraocular: Rara vez puede imitar Rb. Se reportó un caso de un niño de 4 años con leucocoria sospechosa de Rb que fue enucleado, y la patología reveló cisticercosis3)
Para el cribado de niños con Rb familiar, las recomendaciones AAOOP 2018 son ampliamente referenciadas internacionalmente1).
Riesgo
Calendario de cribado
Momento de finalización
Alto (>7.5%)
Nacimiento a 8 semanas: cada 2–4 semanas → 8–12 semanas: mensual → 1–2 años: cada 2 meses → 2–3 años: cada 3 meses → 3–4 años: cada 4 meses → 4–7 años: cada 6 meses
7 años (de por vida para portadores de mutación RB1)
Intermedio (1–7.5%)
Nacimiento a 3 meses: mensual → reducción gradual
7 años
Bajo (<1%)
Nacimiento a 3 meses: una vez al mes → reducción gradual
7 años
Un estudio de cohorte retrospectivo con datos nacionales de los Países Bajos (1991-2019, 38 de 332 pacientes con Rb familiar) mostró que los 28 pacientes que recibieron un cribado completo fueron diagnosticados antes del año de edad (mediana de 18 días), mientras que los 10 pacientes con cribado incompleto tuvieron un diagnóstico significativamente retrasado con una mediana de 420 días (rango 59 días a 4.8 años) 2). También se ha propuesto una revisión del protocolo para reducir la edad de finalización del cribado a 2 años para grupos de bajo riesgo (<3%) 2).
En los casos familiares, la continuación del cribado de fondo de ojo desde inmediatamente después del nacimiento afecta directamente al pronóstico. Los estudios de registro clásicos también muestran que el momento del diagnóstico en los casos familiares está estrechamente relacionado con la frecuencia del cribado, y la práctica actual se está moviendo hacia la individualización del momento de finalización combinando la presencia o ausencia de mutaciones en RB1. 1, 2)
QSi hay antecedentes familiares de retinoblastoma, ¿hasta cuándo es necesario el cribado para el niño?
A
La AAOOP recomienda exámenes regulares de fondo de ojo hasta los 7 años. Con un cribado completo, la mayoría de los casos se diagnostican antes del año de edad. Si las pruebas genéticas descartan el riesgo de mutación en RB1, es posible la finalización temprana del cribado. Para los portadores de mutaciones en RB1, se recomienda un seguimiento irregular cada 1-2 años después de los 7 años.
Para lesiones intraoculares tempranas donde se puede esperar función visual, se realiza activamente un tratamiento de preservación ocular. En etapas intraoculares avanzadas, a menudo no se espera función visual, pero si la familia lo desea, se puede considerar el tratamiento de preservación. El tratamiento requiere un alto nivel de especialización, y es importante la derivación temprana a un centro especializado.
Son elegibles tumores de hasta aproximadamente 3 mm de diámetro. La irradiación directa con láser infrarrojo logra un control local en aproximadamente el 90% de los casos. Si el tumor se encuentra en la mácula, se recomienda quimioterapia sistémica primero para evitar un deterioro visual irreversible.
② Crioterapia
Se tratan tumores de hasta aproximadamente 3 mm en la región ecuatorial o periférica. El método de triple congelación-descongelación, repitiendo la congelación y descongelación tres veces, es común, y se logran tasas de control local de aproximadamente el 90%, similares a la terapia con láser.
③ Braquiterapia
Indicaciones: tumor con grosor ≤5 mm, diámetro mayor ≤15 mm, tumor localizado alejado del disco óptico. En Japón y Europa se utiliza ¹⁰⁶Ru (rutenio-106, fuente beta); en Norteamérica se usan semillas de ¹²⁵I. La fuente radiactiva se sutura temporalmente sobre la esclerótica correspondiente al tumor. Este tratamiento requiere una sala especial y está disponible solo en centros limitados. Se logra un control local del 80–90%.
④ Quimioterapia sistémica (esquema VEC)
Tratamiento de primera línea para tumores intraoculares en etapa avanzada. Se utiliza ampliamente la quimioterapia combinada de tres fármacos, pero la curación solo con quimioterapia es inferior al 10%. Tras la reducción tumoral, se realiza consolidación con tratamientos locales (láser, crioterapia, braquiterapia).
Fármaco
Dosis (basada en superficie corporal)
Dosis (basada en peso para ≤36 meses)
Esquema de administración
Vincristina (Oncovin®)
1.5 mg/m²
0.05 mg/kg
día 1
Carboplatino (Paraplatin®)
560 mg/m²
18.6 mg/kg
día 1
Etopósido (Vepesid®)
150 mg/m²
5 mg/kg
día 1, 2
Repetir cada 3–4 semanas durante 2–6 ciclos (todos por infusión intravenosa).
Se utiliza un catéter para administrar el fármaco (melfalán; inyección de Alkeran®) directamente en la arteria oftálmica. Al administrar una dosis más alta localmente en el ojo y reducir la exposición sistémica, se pueden reducir los efectos secundarios como la supresión de la médula ósea. Aunque no está cubierto por el seguro, es un tratamiento en investigación realizado en más de 20 países del mundo.
Para la siembra vítrea, la quimioterapia sistémica y la infusión intraarterial tienen una eficacia limitada, por lo que se utiliza la inyección intravítrea de melfalán (inyección de Alkeran®) en combinación. Este es un tratamiento en investigación no cubierto por el seguro.
⑦ Radioterapia de haz externo
Irradiación fraccionada con 40–46 Gy de rayos X. Hasta la década de 1990, este fue el pilar del tratamiento de preservación ocular, pero se hicieron evidentes la deformidad del hueso orbitario y el aumento del riesgo de cánceres secundarios. Actualmente, se usa solo cuando otros tratamientos no pueden controlar la enfermedad.
El margen del nervio óptico positivo o la invasión extraescleral son indicaciones absolutas para terapia adyuvante, y se realiza quimioterapia sistémica y radioterapia. La invasión coroidea marcada o la invasión del nervio óptico más allá de la lámina cribosa se evalúan como factores de riesgo relativo de metástasis.
Condiciones para la preservación del ojo
T1 (enfermedad intraocular temprana): Tasa de preservación >90%
Diámetro del tumor ≤3 mm: Terapia láser o criocoagulación como primera opción
Grosor del tumor ≤5 mm: Braquiterapia indicada
Quimioterapia sistémica → tratamiento local: Incluso en casos avanzados, intentar preservación tras reducción
Condiciones que requieren enucleación
T3 (invasión extraocular): Tasa de preservación aproximadamente 10%
Invasión de cámara anterior o iris: Riesgo de diseminación extraocular
Cuando no se espera recuperación de la función visual: Priorizar el pronóstico de vida
Q¿Es posible conservar el ojo?
A
Para tumores intraoculares tempranos (T1), la conservación del ojo es posible en más del 90% de los casos. Se utiliza quimioterapia sistémica (régimen VEC) para reducir el tumor, seguida de consolidación con láser, crioterapia o braquiterapia. En casos avanzados (T3), la tasa de conservación ocular es de aproximadamente el 10%, y puede ser necesaria la enucleación. La elección del tratamiento la determina el especialista según el estadio y la función visual esperada.
El gen RB1, ubicado en el brazo largo del cromosoma 13 (13q14.2), codifica la proteína RB1 (pRb), que desempeña un papel crucial en la regulación de la división celular. pRb se une a los factores de transcripción E2F y suprime la transición G1/S del ciclo celular, controlando así la proliferación celular como proteína supresora de tumores.
Hipótesis de los dos impactos (hipótesis de Knudson)
Según la hipótesis de los dos impactos propuesta por Knudson, la transformación maligna ocurre cuando ambos alelos del gen RB1 en una sola célula están inactivados.
Hereditario: El primer impacto (mutación germinal) está presente en todas las células. Cuando ocurre un segundo impacto (mutación somática, LOH, etc.) en una célula de la retina, se desarrolla el cáncer. Por lo tanto, son comunes los tumores bilaterales y multifocales, y el diagnóstico tiende a ser relativamente temprano.
No hereditario: Tanto el primer como el segundo impacto ocurren como mutaciones somáticas dentro de la misma célula de la retina. Dado que la probabilidad de que ambos impactos coincidan en una sola célula es baja, los tumores suelen ser unilaterales y unifocales, y el diagnóstico tiende a ser más tardío que en los casos hereditarios.
En casos hereditarios, el primer impacto de RB1 está presente en todas las células del cuerpo. Cuando ocurre un segundo impacto en células fuera de la retina (p. ej., hueso, tejido blando), se desarrolla un segundo tumor maligno primario. El osteosarcoma es el más común y a menudo ocurre después de la adolescencia. En casos hereditarios tratados con radioterapia de haz externo, el riesgo de segundos tumores aumenta aún más; por lo tanto, la radioterapia de haz externo ahora se usa solo en situaciones limitadas.
7. Investigación más reciente y perspectivas futuras
La IAC (quimioterapia intraarterial) administra melfalán directamente en la arteria oftálmica, minimizando la toxicidad sistémica mientras se logra una alta concentración intraocular del fármaco. Puede ampliar la oportunidad de preservación ocular incluso en casos avanzados con siembra vítrea. Actualmente se están evaluando los resultados a largo plazo en cohortes a gran escala.
La inyección intravítrea de melfalán se utiliza para la siembra vítrea, pero la estandarización internacional de los protocolos de dosis e intervalo sigue siendo un desafío. Múltiples series de casos han informado altas tasas de control de la siembra, y se espera que futuros ensayos prospectivos establezcan su papel.
Un estudio de cohorte holandés demostró claramente una correlación entre la asistencia completa al cribado y el diagnóstico temprano (mediana de 18 días), cuantificando los efectos adversos de la interrupción del cribado 2). Las propuestas para acortar la edad de finalización del cribado (a 2 años) para grupos de bajo riesgo están avanzando en la optimización del protocolo basada en la estratificación del riesgo 2). La difusión mundial de las recomendaciones de la AAOOP y la estandarización de los protocolos regionales siguen siendo desafíos futuros 1).
Asociación con malformaciones cerebrales congénitas
Aunque raros, existen informes de casos de retinoblastoma que coexisten con malformaciones cerebrales congénitas o anomalías cromosómicas. Un informe de retinoblastoma bilateral con síndrome de Dandy-Walker destaca la importancia de la evaluación sistémica que incluya el antecedente neuroevolutivo, no solo los síntomas oculares. 4)
El uso generalizado de la secuenciación de próxima generación (NGS) ha mejorado la sensibilidad de detección de mutaciones germinales de RB1. También se está estudiando el potencial de la monitorización mediante biopsia líquida (ADN tumoral circulante), y se espera su aplicación para la estratificación pronóstica sin biopsia invasiva.
Se necesita la estandarización de los protocolos de vigilancia del segundo cáncer para los supervivientes a largo plazo de retinoblastoma hereditario. En particular, la frecuencia y el momento de finalización del cribado multiorgánico mediante resonancia magnética de cuerpo entero se están investigando mediante estudios de cohorte continuos para acumular evidencia.
La infusión selectiva de la arteria oftálmica (IAC) y la quimioterapia intravítrea (IVitC) se están estableciendo internacionalmente como tratamientos que mejoran las tasas de preservación ocular en casos avanzados y con siembra vítrea. En las revisiones del tratamiento conservador, se posicionan como estrategias que amplían la preservación ocular sin aumentar el riesgo de metástasis, y el estado de implementación y la centralización de instalaciones especializadas en cada país afectan los resultados. 5)
Disparidades en el tratamiento en países en desarrollo
Mientras que la tasa de supervivencia a 5 años en los países desarrollados supera el 95%, en los países de ingresos bajos y medios de África y Asia se reporta que se sitúa entre el 25 y el 70%. Las revisiones sistemáticas y los análisis por nivel de ingresos del país muestran consistentemente disparidades en las tasas de supervivencia y preservación ocular, siendo los principales factores el diagnóstico tardío, la falta de acceso a la atención médica y la escasez de instalaciones especializadas. La difusión de programas de detección comunitarios, incluida la prueba del reflejo rojo, es un desafío internacional. 6, 7)
Skalet AH, Gombos DS, Gallie BL, Kim JW, Shields CL, Marr BP, Plon SE, Chévez-Barrios P. Screening Children at Risk for Retinoblastoma: Consensus Report from the American Association of Ophthalmic Oncologists and Pathologists. Ophthalmology. 2018;125(3):453-458. doi:10.1016/j.ophtha.2017.09.001. PMID:29056300.
Badalova NA, van Hoefen Wijsard M, Dommering CJ, et al. At what age could screening for familial retinoblastoma be stopped? Revised Dutch retrospective population-based cohort study 1991-2019. Ophthalmology. 2024;131(10):1189-1196.
Jakati S, Vempuluru VS, Kaliki S. Intraocular Cysticercosis Masquerading as Cavitary Retinoblastoma. Ophthalmology. 2025;132(4):e68. doi:10.1016/j.ophtha.2024.05.017. PMID:38878044.
Lomi N, Das D, Chawla B, Parampalli Ravindra A. Retinoblastoma in Dandy-Walker Syndrome. Cureus. 2025;17(8):e89663. doi:10.7759/cureus.89663. PMID:40926918; PMCID:PMC12415497.
Francis L. Munier, Maja Beck-Popovic, Guillermo L. Chantada, David Cobrinik, Tero T. Kivelä, Dietmar Lohmann, Philippe Maeder, Annette C. Moll, et al. Conservative management of retinoblastoma: Challenging orthodoxy without compromising the state of metastatic grace. “Alive, with good vision and no comorbidity”. Progress in Retinal and Eye Research. 2019;73:100764. doi:10.1016/j.preteyeres.2019.05.005.
Wong ES, Choy RW, Zhang Y, et al. Global retinoblastoma survival and globe preservation: a systematic review and meta-analysis. Lancet Glob Health. 2022;10(3):e380-e389.
Global Retinoblastoma Study Group, Fabian ID, Abdallah E, Abdullahi SU, Abdulqader RA, Adamou Boubacar S, Ademola-Popoola DS, Adio A, Afshar AR, Aggarwal P, Aghaji AE, Ahmad A, Akib MNR, Al Harby L, Al Ani MH, Alakbarova A, Portabella SA, Al-Badri SAF, Alcasabas APA, Al-Dahmash SA, Alejos A, Alemany-Rubio E, Alfa Bio AI, Alfonso Carreras Y, Al-Haddad C, Al-Hussaini HHY, Ali AM, Alia DB, Al-Jadiry MF, Al-Jumaily U, Alkatan HM, All-Eriksson C, Al-Mafrachi AARM, Almeida AA, Alsawidi KM, Al-Shaheen AASM, Al-Shammary EH, Amiruddin PO, Antonino R, Astbury NJ, Atalay HT, Atchaneeyasakul LO, Atsiaya R, Attaseth T, Aung TH, Ayala S, Baizakova B, Balaguer J, Balayeva R, Balwierz W, Barranco H, Bascaran C, Beck Popovic M, Benavides R, Benmiloud S, Bennani Guebessi N, Berete RC, Berry JL, Bhaduri A, Bhat S, Biddulph SJ, Biewald EM, Bobrova N, Boehme M, Boldt HC, Bonanomi MTBC, Bornfeld N, Bouda GC, Bouguila H, Boumedane A, Brennan RC, Brichard BG, Buaboonnam J, Calderón-Sotelo P, Calle Jara DA, Camuglia JE, Cano MR, Capra M, Cassoux N, Castela G, Castillo L, Català-Mora J, Chantada GL, Chaudhry S, Chaugule SS, Chauhan A, Chawla B, Chernodrinska VS, Chiwanga FS, Chuluunbat T, Cieslik K, Cockcroft RL, Comsa C, Correa ZM, Correa Llano MG, Corson TW, Cowan-Lyn KE, Csóka M, Cui X, Da Gama IV, Dangboon W, Das A, Das S, Davanzo JM, Davidson A, De Potter P, Delgado KQ, Demirci H, Desjardins L, Diaz Coronado RY, Dimaras H, Dodgshun AJ, Donaldson C, Donato Macedo CR, Dragomir MD, Du Y, Du Bruyn M, Edison KS, Eka Sutyawan IW, El Kettani A, Elbahi AM, Elder JE, Elgalaly D, Elhaddad AM, Elhassan MMA, Elzembely MM, Essuman VA, Evina TGA, Fadoo Z, Fandiño AC, Faranoush M, Fasina O, Fernández DDPG, Fernández-Teijeiro A, Foster A, Frenkel S, Fu LD, Fuentes-Alabi SL, Gallie BL, Gandiwa M, Garcia JL, García Aldana D, Gassant PY, Geel JA, Ghassemi F, Girón AV, Gizachew Z, Goenz MA, Gold AS, Goldberg-Lavid M, Gole GA, Gomel N, Gonzalez E, Gonzalez Perez G, González-Rodríguez L, Garcia Pacheco HN, Graells J, Green L, Gregersen PA, Grigorovski NDAK, Guedenon KM, Gunasekera DS, Gündüz AK, Gupta H, Gupta S, Hadjistilianou T, Hamel P, Hamid SA, Hamzah N, Hansen ED, Harbour JW, Hartnett ME, Hasanreisoglu M, Hassan S, Hassan S, Hederova S, Hernandez J, Hernandez LMC, Hessissen L, Hordofa DF, Huang LC, Hubbard GB, Hummlen M, Husakova K, Hussein Al-Janabi AN, Ida R, Ilic VR, Jairaj V, Jeeva I, Jenkinson H, Ji X, Jo DH, Johnson KP, Johnson WJ, Jones MM, Kabesha TBA, Kabore RL, Kaliki S, Kalinaki A, Kantar M, Kao LY, Kardava T, Kebudi R, Kepak T, Keren-Froim N, Khan ZJ, Khaqan HA, Khauv P, Kheir WJ, Khetan V, Khodabande A, Khotenashvili Z, Kim JW, Kim JH, Kiratli H, Kivelä TT, Klett A, Komba Palet JEK, Krivaitiene D, Kruger M, Kulvichit K, Kuntorini MW, Kyara A, Lachmann ES, Lam CPS, Lam GC, Larson SA, Latinovic S, Laurenti KD, Le BHA, Lecuona K, Leverant AA, Li C, Limbu B, Long QB, López JP, Lukamba RM, Lumbroso L, Luna-Fineman S, Lutfi D, Lysytsia L, Magrath GN, Mahajan A, Majeed AR, Maka E, Makan M, Makimbetov EK, Manda C, Martín Begue N, Mason L, Mason JO, Matende IO, Materin M, Mattosinho CCDS, Matua M, Mayet I, Mbumba FB, McKenzie JD, Medina-Sanson A, Mehrvar A, Mengesha AA, Menon V, Mercado GJVD, Mets MB, Midena E, Mishra DKC, Mndeme FG, Mohamedani AA, Mohammad MT, Moll AC, Montero MM, Morales RA, Moreira C, Mruthyunjaya P, Msina MS, Msukwa G, Mudaliar SS, Muma KI, Munier FL, Murgoi G, Murray TG, Musa KO, Mushtaq A, Mustak H, Muyen OM, Naidu G, Nair AG, Naumenko L, Ndoye Roth PA, Nency YM, Neroev V, Ngo H, Nieves RM, Nikitovic M, Nkanga ED, Nkumbe H, Nuruddin M, Nyaywa M, Obono-Obiang G, Oguego NC, Olechowski A, Oliver SCN, Osei-Bonsu P, Ossandon D, Paez-Escamilla MA, Pagarra H, Painter SL, Paintsil V, Paiva L, Pal BP, Palanivelu MS, Papyan R, Parrozzani R, Parulekar M, Pascual Morales CR, Paton KE, Pawinska-Wasikowska K, Pe’er J, Peña A, Peric S, Pham CTM, Philbert R, Plager DA, Pochop P, Polania RA, Polyakov VG, Pompe MT, Pons JJ, Prat D, Prom V, Purwanto I, Qadir AO, Qayyum S, Qian J, Rahman A, Rahman S, Rahmat J, Rajkarnikar P, Ramanjulu R, Ramasubramanian A, Ramirez-Ortiz MA, Raobela L, Rashid R, Reddy MA, Reich E, Renner LA, Reynders D, Ribadu D, Riheia MM, Ritter-Sovinz P, Rojanaporn D, Romero L, Roy SR, Saab RH, Saakyan S, Sabhan AH, Sagoo MS, Said AMA, Saiju R, Salas B, San Román Pacheco S, Sánchez GL, Sayalith P, Scanlan TA, Schefler AC, Schoeman J, Sedaghat A, Seregard S, Seth R, Shah AS, Shakoor SA, Sharma MK, Sherief ST, Shetye NG, Shields CL, Siddiqui SN, Sidi Cheikh S, Silva S, Singh AD, Singh N, Singh U, Singha P, Sitorus RS, Skalet AH, Soebagjo HD, Sorochynska T, Ssali G, Stacey AW, Staffieri SE, Stahl ED, Stathopoulos C, Stirn Kranjc B, Stones DK, Strahlendorf C, Suarez MEC, Sultana S, Sun X, Sundy M, Superstein R, Supriyadi E, Surukrattanaskul S, Suzuki S, Svojgr K, Sylla F, Tamamyan G, Tan D, Tandili A, Tarrillo Leiva FF, Tashvighi M, Tateshi B, Tehuteru ES, Teixeira LF, Teh KH, Theophile T, Toledano H, Trang DL, Traoré F, Trichaiyaporn S, Tuncer S, Tyau-Tyau H, Umar AB, Unal E, Uner OE, Urbak SF, Ushakova TL, Usmanov RH, Valeina S, van Hoefen Wijsard M, Varadisai A, Vasquez L, Vaughan LO, Veleva-Krasteva NV, Verma N, Victor AA, Viksnins M, Villacís Chafla EG, Vishnevskia-Dai V, Vora T, Wachtel AE, Wackernagel W, Waddell K, Wade PD, Wali AH, Wang YZ, Weiss A, Wilson MW, Wime ADC, Wiwatwongwana A, Wiwatwongwana D, Wolley Dod C, Wongwai P, Xiang D, Xiao Y, Yam JC, Yang H, Yanga JM, Yaqub MA, Yarovaya VA, Yarovoy AA, Ye H, Yousef YA, Yuliawati P, Zapata López AM, Zein E, Zhang C, Zhang Y, Zhao J, Zheng X, Zhilyaeva K, Zia N, Ziko OAO, Zondervan M, Bowman R.. Global Retinoblastoma Presentation and Analysis by National Income Level. JAMA Oncol. 2020;6(5):685-695. doi:10.1001/jamaoncol.2019.6716. PMID:32105305; PMCID:PMC7047856.
Copia el texto del artículo y pégalo en el asistente de IA que prefieras.
Artículo copiado al portapapeles
Abre un asistente de IA abajo y pega el texto copiado en el chat.