Ретинобластома — это злокачественная опухоль сетчатки у младенцев и детей раннего возраста. Она возникает в результате злокачественной трансформации и пролиферации незрелых клеток сетчатки и считается моногенным заболеванием, вызванным мутацией гена RB1, расположенного на длинном плече хромосомы 13 (13q14.2). Гендерных различий нет, 95% случаев диагностируются до 5 лет.
Заболеваемость составляет 1 на 15 000–23 000 рождений, в Японии ежегодно заболевают 70–80 детей. Соотношение односторонних и двусторонних форм составляет 3:2, средний возраст диагностики — 21 месяц для односторонних и 8 месяцев для двусторонних, причем двусторонние диагностируются раньше. В развитых странах пятилетняя выживаемость при локализованной внутриглазной стадии превышает 95%.
Ретинобластома делится на два основных типа в зависимости от типа генетической мутации.
Классификация
Тип мутации
Особенности заболевания
Генетический риск
Наследственная (герминальная мутация)
Герминальная мутация RB1
Часто двусторонние и множественные опухоли
Передается 50% детей
Ненаследственная (соматическая мутация)
Соматическая мутация в одной клетке сетчатки
Односторонняя одиночная опухоль
Не передается следующему поколению
Наследственная (герминальная мутация): Первая мутация присутствует во всех клетках организма. Вторая мутация приводит к раку. Часто возникают двусторонние и множественные опухоли, передается каждому второму ребенку (50%). Риск вторичных раков, таких как остеосаркома (15,7% через 20 лет).
Ненаследственная (соматическая мутация): В одной клетке сетчатки мутируют оба аллеля гена RB1. Возникает односторонняя одиночная опухоль, риск передачи следующему поколению отсутствует.
Однако даже при односторонних случаях некоторые включают герминальные мутации RB1. Не следует отрицать наследственный характер только из-за односторонности; необходимо интерпретировать семейный анамнез, возраст начала заболевания и количество опухолей на основе генетического консультирования и генетической оценки. 1)
Стадирование напрямую связано с тактикой лечения, направленного на сохранение глаза.
Стадия
Состояние поражения
Ориентировочная частота сохранения глаза
T1 (раннее внутриглазное поражение)
Ограничено глазом, без прогрессирования
Более 90%
T2 (прогрессирующее внутриглазное поражение)
Внутриглазное прогрессирование
Около 50%
T3
Прогрессирующее поражение с экстраокулярной инвазией
Около 10%
QНаследуется ли ретинобластома?
A
Примерно 40% случаев являются наследственными (герминальная мутация RB1) и передаются ребенку с вероятностью 50%. Остальные 60% являются ненаследственными (только соматическая мутация) и не несут риска для следующего поколения. Наследственные формы имеют тенденцию к двустороннему и множественному поражению. Рекомендуется проведение генетического тестирования при постановке диагноза и генетическое консультирование.
Aerts I, et al. Retinoblastoma. Orphanet J Rare Dis. 2006 Aug 25; 1:31. Figure 2. PMCID: PMC1586012. License: CC BY.
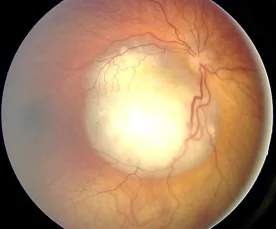
Изображение глазного дна при ретинобластоме, наблюдаемое как богатое сосудами белое возвышающееся образование, демонстрирующее типичную картину, вызывающую лейкокорию. Соответствует находкам на глазном дне и лейкокории, описанным в разделе «2. Основные симптомы и клинические признаки».
Во многих случаях опухоль становится большой внутри глаза и проявляется лейкокорией. При локализации в макуле она может вызывать плохое зрение и косоглазие, что приводит к обнаружению. У детей старшего возраста может отмечаться субъективное снижение зрения; у маленьких детей первым симптомом может быть потирание плохо видящего глаза.
Ранние симптомы
Лейкокория (белый зрачок) : самый частый первый симптом. Опухоль увеличивается внутри глаза, и зрачок выглядит белым.
Косоглазие : вызвано плохим зрением из-за опухоли в макуле. Плохо видящий глаз отклоняется кнаружи.
Субъективное снижение зрения : отмечается у детей старшего возраста.
Потирание глаза : наблюдается у маленьких детей с плохим зрением на пораженном глазу.
Симптомы запущенной стадии
Помутнение роговицы и повышение внутриглазного давления : вызваны сдавлением хрусталика опухолью или неоваскулярной глаукомой (NVG) с повышением внутриглазного давления.
Гиперемия конъюнктивы и отек век : наблюдаются как воспалительные признаки.
Боль: возникает из-за повышения внутриглазного давления или некроза опухоли.
Экзофтальм: наблюдается при экстраокулярном распространении.
Обнаруживается богатое сосудами беловатое возвышающееся образование; при наличии кальцификации постановка окончательного диагноза облегчается. Часто сопровождается диссеминацией в стекловидное тело (распад опухолевых клеток и их рассеивание в стекловидном теле).
Метод красного рефлекса является основой скрининга глазных заболеваний у младенцев. Оценка считается нормальной, если зрачки обоих глаз равны по размеру и дают яркий, симметричный желто-оранжевый рефлекс. Если рефлекс темный или слишком яркий, или имеется разница между глазами, это считается аномалией и требует дальнейшего обследования.
QВсегда ли белый зрачок означает ретинобластому?
A
Причины белого зрачка помимо ретинобластомы включают множество других состояний, таких как персистирующее гиперпластическое первичное стекловидное тело, ретинопатия недоношенных, болезнь Коутса и другие. Однако при обнаружении белого зрачка первоочередной задачей является немедленное обращение к офтальмологу для исключения ретинобластомы. Задержка диагностики напрямую влияет на прогноз, поэтому при подозрении желательно направление в тот же день.
Причиной является мутация гена RB1, расположенного на длинном плече хромосомы 13 (13q14.2). Ген RB1 производит белок RB1 (белок ретинобластомы), который играет важную роль в контроле клеточного деления.
В одной клетке имеется два локуса гена; мутация только в одном локусе сохраняет клеточную функцию, но при мутации обоих локусов клетка теряет способность контролировать деление и становится злокачественной (двухударная теория, гипотеза Кнудсона).
Наследственная форма (герминальная мутация): Первый удар — мутация в зародышевой линии (присутствует во всех клетках). Второй удар происходит в соматической клетке, что приводит к канцерогенезу. Предрасположенность к двусторонним и множественным опухолям.
Ненаследственная форма (соматическая мутация): И первый, и второй удары происходят в одной и той же соматической клетке. Проявляется в виде односторонней одиночной опухоли.
Семейный анамнез является самым большим фактором риска. Определение риска согласно рекомендациям AAOOP (Американской ассоциации офтальмологических онкологов и патологов) приведено ниже1).
Классификация риска
Определение
Значение риска
Высокий
Родитель с двусторонней Rb или носитель герминальной мутации RB1 у родственников первой или второй степени
При наследственных случаях необходимо обращать внимание на риск вторичного рака. Остеосаркома является типичной, возникая у 15,7% наследственных случаев в течение 20 лет. Вторичные раки часто развиваются после 10 лет.
Rumboldt Z, Dodig D, Galluzzi P, et al. Retinoblastoma and beyond: pediatric orbital mass lesions. Neuroradiology. 2025;67(2):469-492. Figure 2. PMID: 39729290; PMCID: PMC11893699; DOI: 10.1007/s00234-024-03517-6. License: CC BY.
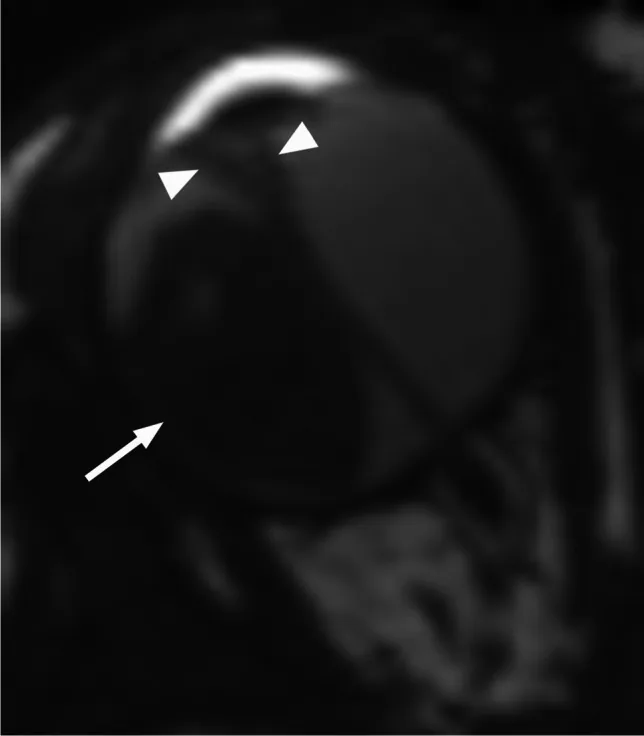
Аксиальная Т2-взвешенная МРТ, показывающая экзофитную ретинобластому (стрелка) с V-образной вторичной отслойкой сетчатки (головка стрелки). Соответствует паттернам роста (экзофитный и эндофитный) и данным МРТ, рассматриваемым в разделе «4. Диагностика и методы обследования».
Биопсия внутриглазной опухоли не проводится. Внутриглазные поражения можно наблюдать непосредственно через прозрачные ткани, и клинический диагноз обладает высокой точностью. Кроме того, биопсия внутриглазной опухоли может привести к экстраокулярному распространению опухолевых клеток, что создает неизбежный риск метастазирования. При глазосохраняющем лечении терапия начинается на основании клинического диагноза.
Офтальмоскопия (основной метод): сосудистое белое возвышающееся образование + кальцификация для клинического подтверждения
УЗИ: подтверждение солидной опухоли и кальцификации. Примечание: у детей старше 5 лет кальцификация выражена слабо.
МРТ: умеренный сигнал на Т1, слегка гипоинтенсивный на Т2, накопление контраста. Обязательно для оценки инфильтрации зрительного нерва и экстраокулярного распространения.
МРТ головы: примерно у 3% двусторонних случаев развивается трилатеральная ретинобластома (опухоль шишковидной железы), поэтому скрининг обязателен.
КТ: отлично визуализирует кальцификаты, но лучевая нагрузка. Если доступна МРТ, КТ имеет вспомогательное значение.
Дифференциальная диагностика заболеваний, вызывающих лейкокорию, имеет первостепенное значение.
Персистирующее фетальное сосудистое образование (персистирующая гиперплазия первичного стекловидного тела): УЗИ для подтверждения наличия или отсутствия солидной опухоли
Ретинопатия недоношенных: Недоношенность и низкая масса тела при рождении в анамнезе являются ключом к дифференциальной диагностике
Болезнь Коутса: Скопление желтовато-белого экссудата под сетчаткой. Сосудистый рисунок опухоли отличается
Астроцитарная гамартома: Дифференциация по наличию сосудов опухоли, локализации на ОКТ и отсутствию роста
Внутриглазной цистицеркоз: Редко, но может имитировать ретинобластому. Сообщается о случае у 4-летнего мальчика с лейкокорией, где после энуклеации по подозрению на Rb патология выявила цистицеркоз3)
Для скрининга детей с семейным Rb широко используется рекомендация AAOOP 20181).
Риск
График скрининга
Возраст завершения
Высокий (>7,5%)
Рождение–8 нед: каждые 2–4 нед → 8–12 нед: ежемесячно → 1–2 года: каждые 2 мес → 2–3 года: каждые 3 мес → 3–4 года: каждые 4 мес → 4–7 лет: каждые 6 мес
7 лет (носители мутации RB1 пожизненно)
Промежуточный (1–7,5%)
Рождение–3 мес: ежемесячно → постепенное снижение
7 лет
Низкий (<1%)
От рождения до 3 месяцев: 1 раз в месяц → постепенное снижение
7 лет
Ретроспективное когортное исследование по данным Нидерландов (1991–2019 гг., 38 семейных случаев Rb из 332) показало, что все 28 пациентов, прошедших полный скрининг, были диагностированы в возрасте до 1 года (медиана 18 дней), тогда как у 10 пациентов с неполным скринингом диагностика была значительно отсрочена (медиана 420 дней, диапазон от 59 дней до 4,8 лет)2). Также было предложено изменить протокол, сократив возраст прекращения скрининга до 2 лет для групп низкого риска (<3%)2).
При семейных случаях продолжение скрининга глазного дна сразу после рождения напрямую влияет на прогноз. В классических регистровых исследованиях время диагностики семейных случаев тесно связано с частотой скрининга, и в настоящее время наблюдается тенденция к индивидуализации сроков прекращения скрининга в зависимости от наличия мутации RB1.1, 2)
QЕсли в семье есть история ретинобластомы, до какого возраста необходимо обследование ребенка?
A
AAOOP рекомендует регулярные осмотры глазного дна до 7 лет. При полном скрининге большинство случаев диагностируется в возрасте до 1 года. Если генетическое тестирование исключает риск мутации RB1, скрининг может быть прекращен раньше. Для носителей мутации RB1 после 7 лет рекомендуется нерегулярное наблюдение каждые 1–2 года.
При ранних внутриглазных поражениях с сохранением зрительной функции активно проводится органосохраняющее лечение. На поздних внутриглазных стадиях зрительная функция часто не сохраняется, но при желании семьи может рассматриваться органосохраняющее лечение. Лечение требует высокой специализации, и раннее направление в специализированный центр имеет решающее значение.
Показано при опухолях диаметром до 3 мм. Прямое облучение инфракрасным лазером обеспечивает локальный контроль примерно в 90% случаев. При опухоли в макулярной области рекомендуется предварительная системная химиотерапия для предотвращения необратимого повреждения зрения.
② Криотерапия
Показана при опухолях размером около 3 мм, расположенных к периферии от экватора. Обычно используется метод тройного замораживания-оттаивания (triple freeze-thaw), который обеспечивает локальный контроль примерно в 90% случаев, аналогично лазеру.
③ Брахитерапия
Показана при локализованных опухолях толщиной ≤ 5 мм и диаметром ≤ 15 мм, расположенных вдали от диска зрительного нерва. В Японии и Европе используется ¹⁰⁶Ru (рутений-106, β-источник), в Северной Америке — источник ¹²⁵I. Лечение заключается во временном подшивании источника к склере над опухолью, что требует специальной процедурной комнаты, поэтому учреждения ограничены. Местный контроль достигается в 80–90% случаев.
④ Системная химиотерапия (схема VEC)
Является терапией первой линии при внутриглазных опухолях в прогрессирующей стадии. Широко используется комбинированная химиотерапия тремя препаратами, но излечение только с помощью этого лечения составляет менее 10%. После уменьшения опухоли проводится локальное лечение (лазер, криотерапия, брахитерапия) для закрепления результата.
Препарат
Доза (на основе площади поверхности тела)
Доза (на основе веса для детей ≤ 36 месяцев)
График введения
Винкристин (Онковин®)
1,5 мг/м²
0,05 мг/кг
День 1
Карбоплатин (Параплатин®)
560 мг/м²
18,6 мг/кг
День 1
Этопозид (Vepesid®)
150 мг/м²
5 мг/кг
день 1, 2
Повторять каждые 3–4 недели 2–6 раз (все внутривенно капельно).
Непосредственное введение препарата (мелфалан; Алкеран® раствор для инъекций) в глазную артерию с помощью катетера. Это позволяет доставить высокую дозу локально в глаз, уменьшая системную дозу и снижая побочные эффекты, такие как миелосупрессия. Хотя это не покрывается страховкой, это экспериментальное лечение проводится более чем в 20 странах.
⑥ Интравитреальная инъекция
При диссеминации в стекловидное тело эффективность системной химиотерапии и артериальной инъекции ограничена, поэтому дополнительно проводится интравитреальная инъекция мелфалана (Алкеран® раствор для инъекций). Это экспериментальное лечение, не покрываемое страховкой.
⑦ Наружная лучевая терапия
Фракционное облучение рентгеновскими лучами в дозе 40–46 Гр. До 1990-х годов это был основной метод сохранения глаза, но стали очевидны деформация орбиты и увеличение вторичных раков, поэтому сейчас его применяют только в случаях, когда другие методы не позволяют контролировать заболевание.
Положительный край зрительного нерва и экстрасклеральная инфильтрация являются абсолютными показаниями к адъювантной терапии, включающей системную химиотерапию и лучевую терапию. Выраженная инфильтрация сосудистой оболочки и инфильтрация зрительного нерва за пределы решетчатой пластинки оцениваются как относительные факторы риска метастазирования.
Условия для сохранения глаза
T1 (раннее внутриглазное поражение) : частота сохранения > 90 %
Диаметр опухоли ≤ 3 мм : лазерная терапия или криокоагуляция как первый выбор
Толщина опухоли ≤ 5 мм : брахитерапия показана
Системная химиотерапия → местное лечение : даже в запущенных случаях попытка сохранения после уменьшения
Условия, требующие энуклеации
T3 (экстраокулярное распространение) : частота сохранения около 10 %
Неоваскулярная глаукома (NVG) : при невозможности контроля внутриглазного давления
Инфильтрация передней камеры или радужки : риск экстраокулярного распространения
Когда восстановление зрительной функции не ожидается : приоритет жизненного прогноза
QВозможно ли сохранить глаз?
A
При ранних внутриглазных поражениях (T1) сохранение глаза возможно более чем в 90% случаев. Системная химиотерапия (VEC-терапия) уменьшает опухоль, затем проводится консолидация с помощью лазера, криотерапии или брахитерапии. При запущенных случаях (T3) частота сохранения глаза составляет около 10%, и может потребоваться энуклеация. Выбор лечения определяется специалистом на основе стадии и прогноза зрительной функции.
Ген RB1, расположенный на длинном плече хромосомы 13 (13q14.2), кодирует белок RB1 (pRb), который играет ключевую роль в контроле клеточного деления. pRb связывается с фактором транскрипции E2F и ингибирует переход G1/S клеточного цикла, действуя как белок-супрессор опухоли, регулирующий клеточную пролиферацию.
Согласно теории двух ударов, предложенной Кнудсоном, злокачественность возникает, когда оба аллеля гена RB1 в одной клетке инактивированы.
Наследственная форма: Первый удар (мутация в зародышевой линии) присутствует во всех клетках. Когда в клетке сетчатки происходит второй удар (соматическая мутация, потеря гетерозиготности и т.д.), развивается рак. Поэтому часто возникают двусторонние и множественные опухоли, и диагноз ставится относительно рано.
Ненаследственная форма: Как первый, так и второй удар возникают в виде соматических мутаций в одной и той же клетке сетчатки. Вероятность случайного совпадения обоих ударов в одной клетке низка, поэтому опухоли часто односторонние и одиночные, а диагноз ставится, как правило, позже, чем при наследственной форме.
При наследственной форме первый удар RB1 присутствует во всех клетках организма. Если второй удар происходит в клетках, отличных от сетчатки (кости, мягкие ткани и т.д.), развивается вторичная первичная злокачественная опухоль. Наиболее частой является остеосаркома, которая часто возникает после 10 лет. У наследственных пациентов, получавших наружную лучевую терапию, риск вторичного рака еще выше, поэтому наружная лучевая терапия в настоящее время используется ограниченно.
IAC (внутриартериальная химиотерапия) заключается в прямом введении мелфалана в глазную артерию, что обеспечивает доставку высокой концентрации препарата в глаз при минимальной системной токсичности. Это может расширить возможности сохранения глаза даже в запущенных случаях с витреальной диссеминацией. В настоящее время продолжается оценка долгосрочных результатов в крупных когортах.
Интравитреальное введение мелфалана проводится для лечения витреальной диссеминации, однако международная стандартизация протоколов дозирования и интервалов остается проблемой. В нескольких сериях случаев сообщается о высоких показателях контроля диссеминации, и будущие проспективные исследования должны установить ее место.
В голландском когортном исследовании была четко продемонстрирована корреляция между прохождением полного скрининга и ранней диагностикой (медиана 18 дней), а также количественно оценены негативные последствия прекращения скрининга 2). Кроме того, продолжается оптимизация протоколов на основе стратификации риска, включая предложения по сокращению возраста прекращения скрининга (2 года) для групп низкого риска 2). Глобальное распространение рекомендаций AAOOP и стандартизация региональных протоколов являются будущими задачами 1).
Сочетание с врожденными пороками развития головного мозга
Хотя и редко, сообщается о случаях ретинобластомы на фоне врожденных пороков развития головного мозга или хромосомных аномалий. Сообщение о двусторонней ретинобластоме с синдромом Денди-Уокера подчеркивает важность системной оценки, включая нейроразвитие, в дополнение к глазным симптомам. 4)
С распространением секвенирования нового поколения (NGS) повысилась чувствительность выявления герминальных мутаций RB1. Также изучается возможность мониторинга с помощью жидкостной биопсии (циркулирующая опухолевая ДНК в крови), и ожидается ее применение для прогностической стратификации без инвазивной биопсии.
Оптимизация наблюдения за вторыми злокачественными новообразованиями
Требуется стандартизация протоколов наблюдения за вторыми злокачественными новообразованиями для долгосрочных выживших после наследственной ретинобластомы. В частности, частота и возраст прекращения мультиорганного скрининга с помощью МРТ всего тела продолжают накапливать доказательную базу в ходе когортных исследований.
Селективная инъекция в глазную артерию (IAC) и интравитреальная химиотерапия (IVitC) становятся международно признанными методами лечения, повышающими показатели сохранения глаза в запущенных случаях или при витреальной диссеминации. В обзорах консервативного лечения они позиционируются как стратегии, расширяющие сохранение глаза без увеличения риска метастазирования, и результаты зависят от внедрения в каждой стране и централизации в специализированных центрах. 5)
В то время как 5-летняя выживаемость в развитых странах превышает 95%, в странах с низким и средним уровнем дохода в Африке и Азии она составляет всего 25–70%. Систематические обзоры и анализ по уровню дохода стран последовательно показывают различия в показателях выживаемости и сохранения глазного яблока, причем основными факторами считаются поздняя диагностика, недостаточный доступ к медицинской помощи и нехватка специализированных учреждений. Распространение программ скрининга на уровне сообществ, включая метод красного рефлекса, является международной задачей. 6, 7)
Skalet AH, Gombos DS, Gallie BL, Kim JW, Shields CL, Marr BP, Plon SE, Chévez-Barrios P. Screening Children at Risk for Retinoblastoma: Consensus Report from the American Association of Ophthalmic Oncologists and Pathologists. Ophthalmology. 2018;125(3):453-458. doi:10.1016/j.ophtha.2017.09.001. PMID:29056300.
Badalova NA, van Hoefen Wijsard M, Dommering CJ, et al. At what age could screening for familial retinoblastoma be stopped? Revised Dutch retrospective population-based cohort study 1991-2019. Ophthalmology. 2024;131(10):1189-1196.
Jakati S, Vempuluru VS, Kaliki S. Intraocular Cysticercosis Masquerading as Cavitary Retinoblastoma. Ophthalmology. 2025;132(4):e68. doi:10.1016/j.ophtha.2024.05.017. PMID:38878044.
Lomi N, Das D, Chawla B, Parampalli Ravindra A. Retinoblastoma in Dandy-Walker Syndrome. Cureus. 2025;17(8):e89663. doi:10.7759/cureus.89663. PMID:40926918; PMCID:PMC12415497.
Francis L. Munier, Maja Beck-Popovic, Guillermo L. Chantada, David Cobrinik, Tero T. Kivelä, Dietmar Lohmann, Philippe Maeder, Annette C. Moll, et al. Conservative management of retinoblastoma: Challenging orthodoxy without compromising the state of metastatic grace. “Alive, with good vision and no comorbidity”. Progress in Retinal and Eye Research. 2019;73:100764. doi:10.1016/j.preteyeres.2019.05.005.
Wong ES, Choy RW, Zhang Y, et al. Global retinoblastoma survival and globe preservation: a systematic review and meta-analysis. Lancet Glob Health. 2022;10(3):e380-e389.
Global Retinoblastoma Study Group, Fabian ID, Abdallah E, Abdullahi SU, Abdulqader RA, Adamou Boubacar S, Ademola-Popoola DS, Adio A, Afshar AR, Aggarwal P, Aghaji AE, Ahmad A, Akib MNR, Al Harby L, Al Ani MH, Alakbarova A, Portabella SA, Al-Badri SAF, Alcasabas APA, Al-Dahmash SA, Alejos A, Alemany-Rubio E, Alfa Bio AI, Alfonso Carreras Y, Al-Haddad C, Al-Hussaini HHY, Ali AM, Alia DB, Al-Jadiry MF, Al-Jumaily U, Alkatan HM, All-Eriksson C, Al-Mafrachi AARM, Almeida AA, Alsawidi KM, Al-Shaheen AASM, Al-Shammary EH, Amiruddin PO, Antonino R, Astbury NJ, Atalay HT, Atchaneeyasakul LO, Atsiaya R, Attaseth T, Aung TH, Ayala S, Baizakova B, Balaguer J, Balayeva R, Balwierz W, Barranco H, Bascaran C, Beck Popovic M, Benavides R, Benmiloud S, Bennani Guebessi N, Berete RC, Berry JL, Bhaduri A, Bhat S, Biddulph SJ, Biewald EM, Bobrova N, Boehme M, Boldt HC, Bonanomi MTBC, Bornfeld N, Bouda GC, Bouguila H, Boumedane A, Brennan RC, Brichard BG, Buaboonnam J, Calderón-Sotelo P, Calle Jara DA, Camuglia JE, Cano MR, Capra M, Cassoux N, Castela G, Castillo L, Català-Mora J, Chantada GL, Chaudhry S, Chaugule SS, Chauhan A, Chawla B, Chernodrinska VS, Chiwanga FS, Chuluunbat T, Cieslik K, Cockcroft RL, Comsa C, Correa ZM, Correa Llano MG, Corson TW, Cowan-Lyn KE, Csóka M, Cui X, Da Gama IV, Dangboon W, Das A, Das S, Davanzo JM, Davidson A, De Potter P, Delgado KQ, Demirci H, Desjardins L, Diaz Coronado RY, Dimaras H, Dodgshun AJ, Donaldson C, Donato Macedo CR, Dragomir MD, Du Y, Du Bruyn M, Edison KS, Eka Sutyawan IW, El Kettani A, Elbahi AM, Elder JE, Elgalaly D, Elhaddad AM, Elhassan MMA, Elzembely MM, Essuman VA, Evina TGA, Fadoo Z, Fandiño AC, Faranoush M, Fasina O, Fernández DDPG, Fernández-Teijeiro A, Foster A, Frenkel S, Fu LD, Fuentes-Alabi SL, Gallie BL, Gandiwa M, Garcia JL, García Aldana D, Gassant PY, Geel JA, Ghassemi F, Girón AV, Gizachew Z, Goenz MA, Gold AS, Goldberg-Lavid M, Gole GA, Gomel N, Gonzalez E, Gonzalez Perez G, González-Rodríguez L, Garcia Pacheco HN, Graells J, Green L, Gregersen PA, Grigorovski NDAK, Guedenon KM, Gunasekera DS, Gündüz AK, Gupta H, Gupta S, Hadjistilianou T, Hamel P, Hamid SA, Hamzah N, Hansen ED, Harbour JW, Hartnett ME, Hasanreisoglu M, Hassan S, Hassan S, Hederova S, Hernandez J, Hernandez LMC, Hessissen L, Hordofa DF, Huang LC, Hubbard GB, Hummlen M, Husakova K, Hussein Al-Janabi AN, Ida R, Ilic VR, Jairaj V, Jeeva I, Jenkinson H, Ji X, Jo DH, Johnson KP, Johnson WJ, Jones MM, Kabesha TBA, Kabore RL, Kaliki S, Kalinaki A, Kantar M, Kao LY, Kardava T, Kebudi R, Kepak T, Keren-Froim N, Khan ZJ, Khaqan HA, Khauv P, Kheir WJ, Khetan V, Khodabande A, Khotenashvili Z, Kim JW, Kim JH, Kiratli H, Kivelä TT, Klett A, Komba Palet JEK, Krivaitiene D, Kruger M, Kulvichit K, Kuntorini MW, Kyara A, Lachmann ES, Lam CPS, Lam GC, Larson SA, Latinovic S, Laurenti KD, Le BHA, Lecuona K, Leverant AA, Li C, Limbu B, Long QB, López JP, Lukamba RM, Lumbroso L, Luna-Fineman S, Lutfi D, Lysytsia L, Magrath GN, Mahajan A, Majeed AR, Maka E, Makan M, Makimbetov EK, Manda C, Martín Begue N, Mason L, Mason JO, Matende IO, Materin M, Mattosinho CCDS, Matua M, Mayet I, Mbumba FB, McKenzie JD, Medina-Sanson A, Mehrvar A, Mengesha AA, Menon V, Mercado GJVD, Mets MB, Midena E, Mishra DKC, Mndeme FG, Mohamedani AA, Mohammad MT, Moll AC, Montero MM, Morales RA, Moreira C, Mruthyunjaya P, Msina MS, Msukwa G, Mudaliar SS, Muma KI, Munier FL, Murgoi G, Murray TG, Musa KO, Mushtaq A, Mustak H, Muyen OM, Naidu G, Nair AG, Naumenko L, Ndoye Roth PA, Nency YM, Neroev V, Ngo H, Nieves RM, Nikitovic M, Nkanga ED, Nkumbe H, Nuruddin M, Nyaywa M, Obono-Obiang G, Oguego NC, Olechowski A, Oliver SCN, Osei-Bonsu P, Ossandon D, Paez-Escamilla MA, Pagarra H, Painter SL, Paintsil V, Paiva L, Pal BP, Palanivelu MS, Papyan R, Parrozzani R, Parulekar M, Pascual Morales CR, Paton KE, Pawinska-Wasikowska K, Pe’er J, Peña A, Peric S, Pham CTM, Philbert R, Plager DA, Pochop P, Polania RA, Polyakov VG, Pompe MT, Pons JJ, Prat D, Prom V, Purwanto I, Qadir AO, Qayyum S, Qian J, Rahman A, Rahman S, Rahmat J, Rajkarnikar P, Ramanjulu R, Ramasubramanian A, Ramirez-Ortiz MA, Raobela L, Rashid R, Reddy MA, Reich E, Renner LA, Reynders D, Ribadu D, Riheia MM, Ritter-Sovinz P, Rojanaporn D, Romero L, Roy SR, Saab RH, Saakyan S, Sabhan AH, Sagoo MS, Said AMA, Saiju R, Salas B, San Román Pacheco S, Sánchez GL, Sayalith P, Scanlan TA, Schefler AC, Schoeman J, Sedaghat A, Seregard S, Seth R, Shah AS, Shakoor SA, Sharma MK, Sherief ST, Shetye NG, Shields CL, Siddiqui SN, Sidi Cheikh S, Silva S, Singh AD, Singh N, Singh U, Singha P, Sitorus RS, Skalet AH, Soebagjo HD, Sorochynska T, Ssali G, Stacey AW, Staffieri SE, Stahl ED, Stathopoulos C, Stirn Kranjc B, Stones DK, Strahlendorf C, Suarez MEC, Sultana S, Sun X, Sundy M, Superstein R, Supriyadi E, Surukrattanaskul S, Suzuki S, Svojgr K, Sylla F, Tamamyan G, Tan D, Tandili A, Tarrillo Leiva FF, Tashvighi M, Tateshi B, Tehuteru ES, Teixeira LF, Teh KH, Theophile T, Toledano H, Trang DL, Traoré F, Trichaiyaporn S, Tuncer S, Tyau-Tyau H, Umar AB, Unal E, Uner OE, Urbak SF, Ushakova TL, Usmanov RH, Valeina S, van Hoefen Wijsard M, Varadisai A, Vasquez L, Vaughan LO, Veleva-Krasteva NV, Verma N, Victor AA, Viksnins M, Villacís Chafla EG, Vishnevskia-Dai V, Vora T, Wachtel AE, Wackernagel W, Waddell K, Wade PD, Wali AH, Wang YZ, Weiss A, Wilson MW, Wime ADC, Wiwatwongwana A, Wiwatwongwana D, Wolley Dod C, Wongwai P, Xiang D, Xiao Y, Yam JC, Yang H, Yanga JM, Yaqub MA, Yarovaya VA, Yarovoy AA, Ye H, Yousef YA, Yuliawati P, Zapata López AM, Zein E, Zhang C, Zhang Y, Zhao J, Zheng X, Zhilyaeva K, Zia N, Ziko OAO, Zondervan M, Bowman R.. Global Retinoblastoma Presentation and Analysis by National Income Level. JAMA Oncol. 2020;6(5):685-695. doi:10.1001/jamaoncol.2019.6716. PMID:32105305; PMCID:PMC7047856.
Скопируйте текст статьи и вставьте его в выбранный ИИ-ассистент.
Статья скопирована в буфер обмена
Откройте ИИ-ассистент ниже и вставьте скопированный текст в чат.