Il retinoblastoma è un tumore maligno della retina nei neonati e nei bambini piccoli. Deriva dalla trasformazione maligna e dalla proliferazione di cellule retiniche immature ed è considerato una malattia monogenica causata da una mutazione del gene RB1 situato sul braccio lungo del cromosoma 13 (13q14.2). Non c’è differenza di sesso e il 95% dei casi viene diagnosticato entro i 5 anni di età.
L’incidenza è di 1 ogni 15.000-23.000 nati vivi, con 70-80 nuovi casi all’anno in Giappone. Il rapporto tra forme unilaterali e bilaterali è di 3:2, l’età media alla diagnosi è di 21 mesi per le unilaterali e di 8 mesi per le bilaterali, che vengono diagnosticate più precocemente. Nei paesi sviluppati, il tasso di sopravvivenza a 5 anni per lo stadio intraoculare localizzato supera il 95%.
Il retinoblastoma è suddiviso in due grandi categorie in base al tipo di mutazione genetica.
Classificazione
Tipo di mutazione
Caratteristiche patologiche
Rischio genetico
Ereditario (mutazione germinale)
Mutazione RB1 nella linea germinale
Frequente bilateralità e tumori multipli
Trasmesso al 50% dei figli
Non ereditario (mutazione somatica)
Mutazione somatica in una cellula retinica
Tumore unilaterale e singolo
Nessuna trasmissione alla prole
Ereditario (mutazione germinale): Tutte le cellule del corpo presentano la prima mutazione. La seconda mutazione porta al cancro. Tendenza a tumori bilaterali e multipli, trasmesso a 1 bambino su 2 (50%). Rischio di tumori secondari come l’osteosarcoma (15,7% a 20 anni).
Non ereditario (mutazione somatica): Entrambi gli alleli del gene RB1 sono mutati in una singola cellula retinica. Tumore unilaterale e singolo, nessun rischio di trasmissione alla prole.
Tuttavia, anche nei casi unilaterali, alcuni includono mutazioni germinali di RB1. Non si deve negare l’ereditarietà solo perché unilaterale; è necessario interpretare la storia familiare, l’età di insorgenza e il numero di tumori sulla base di consulenza genetica e valutazione genetica. 1)
Classificazione per stadio e tasso di conservazione del bulbo oculare
La classificazione per stadio è direttamente collegata alla strategia di trattamento per la conservazione del bulbo oculare.
Stadio
Stato della lesione
Tasso indicativo di conservazione del bulbo oculare
T1 (lesione intraoculare precoce)
Limitato all’occhio, senza progressione
Oltre il 90%
T2 (lesione intraoculare avanzata)
Progressione intraoculare
Circa il 50%
T3
Lesione avanzata con invasione extraoculare
Circa il 10%
QIl retinoblastoma è ereditario?
A
Circa il 40% dei casi è ereditario (mutazione germinale RB1) e viene trasmesso al bambino con una probabilità del 50%. Il restante 60% circa è non ereditario (solo mutazione somatica) e non comporta rischio per la generazione successiva. Le forme ereditarie tendono a essere bilaterali e multifocali. Al momento della diagnosi si raccomanda di eseguire un test genetico e di ricevere una consulenza genetica.
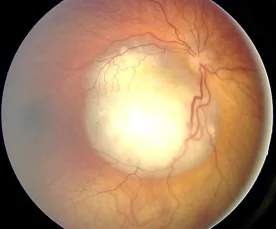
Aerts I, et al. Retinoblastoma. Orphanet J Rare Dis. 2006 Aug 25; 1:31. Figure 2. PMCID: PMC1586012. License: CC BY.
Immagine del fondo oculare di un retinoblastoma osservato come una lesione bianca rilevata riccamente vascolarizzata, che mostra l’aspetto tipico responsabile della leucocoria. Corrisponde ai reperti del fondo oculare e alla leucocoria trattati nella sezione «2. Principali sintomi e segni clinici».
In molti casi il tumore diventa grande all’interno dell’occhio e si manifesta con leucocoria. Quando si sviluppa nella macula, può causare scarsa visione e strabismo, portando alla scoperta. Nei bambini più grandi può essere avvertita una riduzione dell’acuità visiva; nei bambini piccoli, il gesto di strofinare l’occhio con scarsa visione può essere il primo sintomo.
Sintomi precoci
Leucocoria (pupilla bianca) : sintomo iniziale più frequente. Il tumore si ingrandisce nell’occhio e la pupilla appare bianca.
Strabismo : causato dalla scarsa visione dovuta a un tumore maculare. L’occhio con scarsa visione devia verso l’esterno.
Riduzione soggettiva della vista : osservata nei bambini più grandi.
Strofinamento dell’occhio : osservato nei bambini piccoli con scarsa visione nell’occhio interessato.
Sintomi in fase avanzata
Opacità corneale e aumento della pressione intraoculare : causati dalla compressione del cristallino da parte del tumore o da glaucoma neovascolare (NVG) con aumento della pressione intraoculare.
Iperemia congiuntivale e gonfiore palpebrale : osservati come segni infiammatori.
Dolore: compare a causa dell’aumento della pressione intraoculare o della necrosi tumorale.
Es oftalmo: si osserva in caso di estensione extraoculare.
Si osserva una lesione biancastra, rilevata e ricca di vasi; la presenza di calcificazioni facilita la diagnosi definitiva. Spesso si accompagna a disseminazione vitreale (dispersione di cellule tumorali nel vitreo).
Il test del riflesso rosso è la base dello screening delle malattie oculari nei neonati. La valutazione è normale se le pupille di entrambi gli occhi sono di dimensioni uguali e presentano un riflesso giallo-arancio brillante e simmetrico. Se il riflesso è scuro o troppo luminoso, o se c’è una differenza tra i due occhi, è necessaria un’indagine approfondita.
QUna pupilla bianca significa sempre retinoblastoma?
A
Le cause di pupilla bianca includono, oltre al retinoblastoma, molte altre condizioni come la persistenza del vitreo primitivo iperplastico, la retinopatia del prematuro, la malattia di Coats, ecc. Tuttavia, in presenza di pupilla bianca, la priorità assoluta è una rapida visita oculistica per escludere il retinoblastoma. Il ritardo diagnostico influisce direttamente sulla prognosi, quindi in caso di sospetto è opportuno un invio immediato.
La causa è una mutazione del gene RB1 situato sul braccio lungo del cromosoma 13 (13q14.2). Il gene RB1 produce la proteina RB1 (proteina del retinoblastoma), che svolge un ruolo importante nel controllo della divisione cellulare.
Una cellula possiede due loci genici; una mutazione in un solo locus mantiene la funzione cellulare, ma quando entrambi i loci sono mutati, la cellula perde il controllo della divisione e diventa maligna (teoria dei due colpi, ipotesi di Knudson).
Forma ereditaria (mutazione germinale): Il primo colpo è una mutazione della linea germinale (presente in tutte le cellule). Il secondo colpo si verifica in una cellula somatica, portando alla cancerogenesi. Predisposizione a tumori bilaterali e multipli.
Forma non ereditaria (mutazione somatica): Sia il primo che il secondo colpo si verificano nella stessa cellula somatica. Si presenta come tumore unilaterale e singolo.
La storia familiare è il più grande fattore di rischio. La definizione di rischio secondo le raccomandazioni dell’AAOOP (American Association of Ophthalmic Oncologists and Pathologists) è riportata di seguito1).
Classificazione del rischio
Definizione
Valore di rischio
Alto
Genitore con Rb bilaterale o portatore di mutazione germinale RB1 in parenti di primo o secondo grado
Nei casi ereditari è necessario prestare attenzione al rischio di secondo tumore. L’osteosarcoma è tipico e si verifica nel 15,7% dei casi ereditari entro 20 anni. I secondi tumori si manifestano spesso dopo i 10 anni di età.
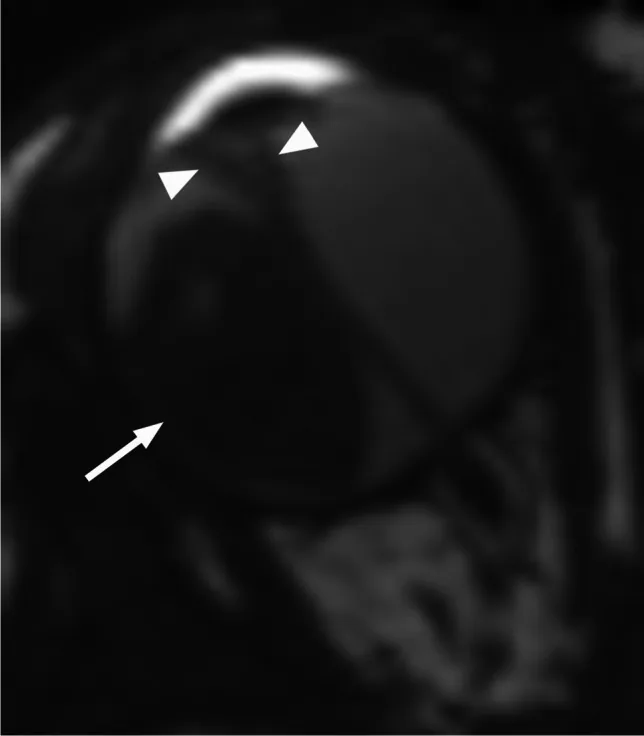
Rumboldt Z, Dodig D, Galluzzi P, et al. Retinoblastoma and beyond: pediatric orbital mass lesions. Neuroradiology. 2025;67(2):469-492. Figure 2. PMID: 39729290; PMCID: PMC11893699; DOI: 10.1007/s00234-024-03517-6. License: CC BY.
RM assiale pesata in T2 che mostra un retinoblastoma esofitico (freccia) con distacco retinico secondario a forma di V (punta di freccia). Corrisponde ai pattern di crescita esofitico ed endofitico e ai reperti RM discussi nella sezione «4. Diagnosi e metodi di esame».
La biopsia di un tumore intraoculare non viene eseguita. Le lesioni intraoculari possono essere osservate direttamente attraverso tessuti trasparenti e l’accuratezza della diagnosi clinica è elevata. Inoltre, la biopsia di un tumore intraoculare può causare la disseminazione extraoculare di cellule tumorali, con un rischio inevitabile di metastasi. In caso di trattamento conservativo dell’occhio, la terapia viene iniziata sulla base della diagnosi clinica.
Esame del fondo oculare (principale): lesione bianca rilevata ricca di vasi + calcificazione per diagnosi clinica definitiva
Ecografia: conferma di tumore solido e calcificazioni. Attenzione: la calcificazione è scarsa nei bambini di età superiore a 5 anni.
RM: segnale moderato in T1, lievemente ipointenso in T2, enhancement dopo contrasto. Essenziale per valutare l’infiltrazione del nervo ottico e l’estensione extraoculare.
RM cranica: circa il 3% dei casi bilaterali sviluppa retinoblastoma trilaterale (tumore pineale), quindi lo screening è obbligatorio.
TC: eccellente per visualizzare le calcificazioni ma esposizione a radiazioni. Se la RM è disponibile, la TC è ausiliaria.
La diagnosi differenziale delle malattie che causano leucocoria è di fondamentale importanza.
Persistenza di residui vascolari fetali (iperplasia persistente del vitreo primario): L’ecografia conferma la presenza o assenza di tumore solido
Retinopatia del prematuro: Anamnesi di parto pretermine e basso peso alla nascita sono indizi per la diagnosi differenziale
Malattia di Coats: Accumulo di essudato giallo-biancastro sottoretinico. Il pattern vascolare tumorale è diverso
Amartoma astrocitario: La diagnosi differenziale si basa sulla presenza di vasi tumorali, localizzazione all’OCT e assenza di crescita
Cisticercosi intraoculare: Rara, ma può mimare il retinoblastoma. Segnalato un caso di un bambino di 4 anni con leucocoria, enucleato per sospetto Rb, con diagnosi patologica di cisticercosi3)
Per lo screening dei bambini con Rb familiare, la raccomandazione AAOOP 2018 è ampiamente utilizzata a livello internazionale1).
Rischio
Programma di screening
Età di conclusione
Alto (>7,5%)
Nascita–8 sett: ogni 2–4 sett → 8–12 sett: mensile → 1–2 anni: ogni 2 mesi → 2–3 anni: ogni 3 mesi → 3–4 anni: ogni 4 mesi → 4–7 anni: ogni 6 mesi
7 anni (portatori di mutazione RB1: a vita)
Intermedio (1–7,5%)
Nascita–3 mesi: mensile → riduzione graduale
7 anni
Basso (<1%)
Dalla nascita a 3 mesi: 1 volta/mese → riduzione graduale
7 anni
Uno studio di coorte retrospettivo nazionale olandese (1991-2019, 38 casi familiari su 332) ha mostrato che tutti i 28 pazienti sottoposti a screening completo sono stati diagnosticati entro il primo anno di vita (mediana 18 giorni), mentre nei 10 pazienti con screening incompleto la diagnosi è stata notevolmente ritardata (mediana 420 giorni, range 59 giorni - 4,8 anni)2). È stata anche proposta una modifica del protocollo per ridurre l’età di fine screening a 2 anni per i gruppi a basso rischio (<3%)2).
Nei casi familiari, la continuazione dello screening del fondo oculare subito dopo la nascita ha un impatto diretto sulla prognosi. Anche negli studi di registro classici, il momento della diagnosi nei casi familiari è strettamente correlato alla frequenza dello screening e attualmente si sta procedendo verso l’individualizzazione del termine dello screening in base alla presenza o assenza di mutazione RB1.1, 2)
QSe in famiglia c'è una storia di retinoblastoma, fino a quando è necessario lo screening del bambino?
A
L’AAOOP raccomanda esami regolari del fondo oculare fino all’età di 7 anni. Con uno screening completo, la maggior parte dei casi viene diagnosticata entro il primo anno di vita. Se il test genetico esclude il rischio di mutazione RB1, lo screening può essere terminato prima. Per i portatori di mutazione RB1, dopo i 7 anni si raccomanda un follow-up irregolare ogni 1-2 anni.
Per le lesioni intraoculari precoci con funzione visiva preservata, si esegue attivamente un trattamento di conservazione dell’occhio. Negli stadi intraoculari avanzati, la funzione visiva è spesso compromessa, ma su richiesta della famiglia si può prendere in considerazione un trattamento conservativo. Il trattamento richiede un’elevata specializzazione ed è importante un rinvio precoce a un centro specializzato.
Indicato per tumori fino a circa 3 mm di diametro. L’irradiazione diretta con laser a infrarossi consente un controllo locale di circa il 90%. In caso di tumore maculare, si raccomanda una chemioterapia sistemica preliminare per evitare danni visivi irreversibili.
② Crioterapia
Indicata per tumori di circa 3 mm situati alla periferia dell’equatore. La tecnica del triplo congelamento-scongelamento (triple freeze-thaw) è comune e consente un controllo locale di circa il 90%, simile al laser.
③ Brachiterapia
È indicata per tumori localizzati, distanti dalla papilla ottica, con spessore ≤ 5 mm e diametro ≤ 15 mm. In Giappone e in Europa si utilizza ¹⁰⁶Ru (rutenio-106, sorgente β), mentre in Nord America si utilizza una sorgente di ¹²⁵I. Il trattamento consiste nel suturare temporaneamente la sorgente sulla sclera in corrispondenza del tumore, richiedendo una sala di trattamento speciale, il che limita le strutture. È possibile un controllo locale dell’80-90%.
④ Chemioterapia sistemica (schema VEC)
È il trattamento di prima linea per i tumori intraoculari in stadio avanzato. Viene ampiamente utilizzata una chemioterapia combinata con tre farmaci, ma il tasso di guarigione con il solo trattamento è inferiore al 10%. Dopo la riduzione del tumore, si esegue un trattamento locale (laser, crioterapia, brachiterapia) per consolidare.
Farmaco
Dose (basata sulla superficie corporea)
Dose (basata sul peso per ≤ 36 mesi)
Schema di somministrazione
Vincristina (Oncovin®)
1,5 mg/m²
0,05 mg/kg
Giorno 1
Carboplatino (Paraplatino®)
560 mg/m²
18,6 mg/kg
Giorno 1
Etoposide (Vepesid®)
150 mg/m²
5 mg/kg
giorno 1, 2
Ripetere ogni 3-4 settimane per 2-6 volte (tutti per infusione endovenosa).
Somministrazione diretta di un farmaco (melfalan; Alkeran® soluzione iniettabile) nell’arteria oftalmica tramite catetere. Ciò consente di somministrare una dose elevata localmente nell’occhio, riducendo la dose sistemica e diminuendo gli effetti collaterali come la mielosoppressione. Sebbene non coperto dall’assicurazione sanitaria, è un trattamento sperimentale praticato in oltre 20 paesi.
Per la disseminazione vitreale, l’efficacia della chemioterapia sistemica e dell’iniezione arteriosa è limitata, pertanto si associa un’iniezione intravitreale di melfalan (Alkeran® soluzione iniettabile). Si tratta di un trattamento sperimentale non coperto dall’assicurazione sanitaria.
⑦ Radioterapia esterna
Irradiazione frazionata con raggi X da 40 a 46 Gy. Fino agli anni ‘90 era il cardine del trattamento conservativo dell’occhio, ma sono diventate evidenti la deformazione orbitaria e l’aumento dei tumori secondari, e ora viene utilizzata solo quando altri trattamenti non consentono il controllo.
La positività del margine del nervo ottico e l’infiltrazione extrasclerale sono indicazioni assolute per la terapia adiuvante, che comprende chemioterapia sistemica e radioterapia. L’infiltrazione coroidale marcata e l’infiltrazione del nervo ottico oltre la lamina cribrosa sono valutate come fattori di rischio relativi per metastasi.
Infiltrazione della camera anteriore o dell’iride : rischio di disseminazione extraoculare
Quando non è previsto recupero della funzione visiva : priorità alla prognosi vitale
QÈ possibile conservare l'occhio?
A
Per le lesioni intraoculari precoci (T1), la conservazione dell’occhio è possibile in oltre il 90% dei casi. La chemioterapia sistemica (VEC) riduce il tumore, seguita da un trattamento di consolidamento con laser, crioterapia o brachiterapia. Nei casi avanzati (T3), il tasso di conservazione dell’occhio è di circa il 10% e può essere necessaria l’enucleazione. La scelta del trattamento è determinata da uno specialista in base allo stadio e alla prognosi della funzione visiva.
Il gene RB1 situato sul braccio lungo del cromosoma 13 (13q14.2) codifica per la proteina RB1 (pRb), che svolge un ruolo chiave nel controllo della divisione cellulare. pRb si lega al fattore di trascrizione E2F e inibisce la transizione G1/S del ciclo cellulare, agendo come una proteina oncosoppressore che regola la proliferazione cellulare.
Secondo la teoria dei due colpi proposta da Knudson, la malignità si verifica quando entrambi gli alleli del gene RB1 in una cellula vengono inattivati.
Ereditario: Il primo colpo (mutazione germinale) è presente in tutte le cellule. Quando si verifica un secondo colpo (mutazione somatica, perdita di eterozigosi, ecc.) in una cellula retinica, si sviluppa il cancro. Pertanto, sono frequenti tumori bilaterali e multipli, e la diagnosi è relativamente precoce.
Non ereditario: Sia il primo che il secondo colpo si verificano come mutazioni somatiche nella stessa cellula retinica. La probabilità che entrambi i colpi si verifichino casualmente in una cellula è bassa, quindi i tumori sono spesso unilaterali e singoli, e la diagnosi tende a essere più tardiva rispetto ai casi ereditari.
Nei casi ereditari, il primo colpo di RB1 è presente in tutte le cellule del corpo. Se un secondo colpo si verifica in cellule diverse dalla retina (osso, tessuti molli, ecc.), si sviluppa un tumore maligno primario secondario. L’osteosarcoma è il più frequente e si verifica spesso dopo i 10 anni. Nei pazienti ereditari che hanno ricevuto radioterapia esterna, il rischio di tumore secondario è ulteriormente aumentato, pertanto la radioterapia esterna è ora utilizzata in modo limitato.
L’IAC (chemioterapia intra-arteriosa) consiste nell’iniettare melphalan direttamente nell’arteria oftalmica, consentendo la somministrazione di farmaci ad alta concentrazione all’interno dell’occhio riducendo al minimo la tossicità sistemica. Può ampliare le possibilità di conservazione del bulbo oculare anche nei casi avanzati con disseminazione vitreale. Attualmente è in corso la valutazione a lungo termine dei risultati in ampie coorti.
L’iniezione intravitreale di melphalan viene eseguita per trattare la disseminazione vitreale, ma la standardizzazione internazionale dei protocolli di dosaggio e intervallo di somministrazione rimane una sfida. Diverse serie di casi hanno riportato alti tassi di controllo della disseminazione e si prevede che futuri studi prospettici ne stabiliranno il ruolo.
In uno studio di coorte olandese, è stata chiaramente dimostrata una correlazione tra la partecipazione allo screening completo e la diagnosi precoce (mediana 18 giorni), e gli effetti negativi dell’interruzione dello screening sono stati quantificati 2). Inoltre, l’ottimizzazione dei protocolli basata sulla stratificazione del rischio sta progredendo, comprese proposte per ridurre l’età di fine dello screening (2 anni) per i gruppi a basso rischio 2). La diffusione globale delle raccomandazioni AAOOP e la standardizzazione dei protocolli regionali sono sfide future 1).
Associazione con malformazioni cerebrali congenite
Sebbene rari, sono stati riportati casi di retinoblastoma in associazione con malformazioni cerebrali congenite o anomalie cromosomiche. Un rapporto di retinoblastoma bilaterale con sindrome di Dandy-Walker sottolinea l’importanza di una valutazione sistemica che includa il background neuroevolutivo oltre ai sintomi oculari. 4)
Con la diffusione del sequenziamento di nuova generazione (NGS), la sensibilità di rilevamento delle mutazioni germinali di RB1 è migliorata. È anche in fase di studio la possibilità di monitoraggio tramite biopsia liquida (DNA tumorale circolante nel sangue) e si prevede la sua applicazione per la stratificazione prognostica evitando biopsie invasive.
Ottimizzazione della sorveglianza per tumori secondari
È necessaria la standardizzazione dei protocolli di sorveglianza per tumori secondari nei sopravvissuti a lungo termine con retinoblastoma ereditario. In particolare, la frequenza e l’età di termine dello screening multi-organo mediante RM total body sono oggetto di accumulo continuo di evidenze attraverso studi di coorte.
L’iniezione arteriosa oculare selettiva (IAC) e la chemioterapia intravitreale (IVitC) si stanno affermando a livello internazionale come trattamenti che hanno aumentato i tassi di conservazione del bulbo oculare nei casi avanzati o con disseminazione vitreale. Nelle revisioni sul trattamento conservativo, sono posizionate come strategie che ampliano la conservazione dell’occhio senza aumentare il rischio metastatico, e i risultati sono influenzati dallo stato di adozione in ciascun paese e dalla centralizzazione in centri specializzati. 5)
Disparità di trattamento nei paesi in via di sviluppo
Mentre il tasso di sopravvivenza a 5 anni nei paesi sviluppati supera il 95%, nei paesi a basso e medio reddito dell’Africa e dell’Asia si attesta solo tra il 25 e il 70%. Revisioni sistematiche e analisi per livello di reddito nazionale mostrano costantemente disparità nei tassi di sopravvivenza e di conservazione del bulbo oculare, con diagnosi tardiva, accesso insufficiente alle cure e mancanza di strutture specializzate come fattori principali. La diffusione di programmi di screening basati sulla comunità, incluso il metodo del riflesso rosso, è una sfida internazionale. 6, 7)
Skalet AH, Gombos DS, Gallie BL, Kim JW, Shields CL, Marr BP, Plon SE, Chévez-Barrios P. Screening Children at Risk for Retinoblastoma: Consensus Report from the American Association of Ophthalmic Oncologists and Pathologists. Ophthalmology. 2018;125(3):453-458. doi:10.1016/j.ophtha.2017.09.001. PMID:29056300.
Badalova NA, van Hoefen Wijsard M, Dommering CJ, et al. At what age could screening for familial retinoblastoma be stopped? Revised Dutch retrospective population-based cohort study 1991-2019. Ophthalmology. 2024;131(10):1189-1196.
Jakati S, Vempuluru VS, Kaliki S. Intraocular Cysticercosis Masquerading as Cavitary Retinoblastoma. Ophthalmology. 2025;132(4):e68. doi:10.1016/j.ophtha.2024.05.017. PMID:38878044.
Lomi N, Das D, Chawla B, Parampalli Ravindra A. Retinoblastoma in Dandy-Walker Syndrome. Cureus. 2025;17(8):e89663. doi:10.7759/cureus.89663. PMID:40926918; PMCID:PMC12415497.
Francis L. Munier, Maja Beck-Popovic, Guillermo L. Chantada, David Cobrinik, Tero T. Kivelä, Dietmar Lohmann, Philippe Maeder, Annette C. Moll, et al. Conservative management of retinoblastoma: Challenging orthodoxy without compromising the state of metastatic grace. “Alive, with good vision and no comorbidity”. Progress in Retinal and Eye Research. 2019;73:100764. doi:10.1016/j.preteyeres.2019.05.005.
Wong ES, Choy RW, Zhang Y, et al. Global retinoblastoma survival and globe preservation: a systematic review and meta-analysis. Lancet Glob Health. 2022;10(3):e380-e389.
Global Retinoblastoma Study Group, Fabian ID, Abdallah E, Abdullahi SU, Abdulqader RA, Adamou Boubacar S, Ademola-Popoola DS, Adio A, Afshar AR, Aggarwal P, Aghaji AE, Ahmad A, Akib MNR, Al Harby L, Al Ani MH, Alakbarova A, Portabella SA, Al-Badri SAF, Alcasabas APA, Al-Dahmash SA, Alejos A, Alemany-Rubio E, Alfa Bio AI, Alfonso Carreras Y, Al-Haddad C, Al-Hussaini HHY, Ali AM, Alia DB, Al-Jadiry MF, Al-Jumaily U, Alkatan HM, All-Eriksson C, Al-Mafrachi AARM, Almeida AA, Alsawidi KM, Al-Shaheen AASM, Al-Shammary EH, Amiruddin PO, Antonino R, Astbury NJ, Atalay HT, Atchaneeyasakul LO, Atsiaya R, Attaseth T, Aung TH, Ayala S, Baizakova B, Balaguer J, Balayeva R, Balwierz W, Barranco H, Bascaran C, Beck Popovic M, Benavides R, Benmiloud S, Bennani Guebessi N, Berete RC, Berry JL, Bhaduri A, Bhat S, Biddulph SJ, Biewald EM, Bobrova N, Boehme M, Boldt HC, Bonanomi MTBC, Bornfeld N, Bouda GC, Bouguila H, Boumedane A, Brennan RC, Brichard BG, Buaboonnam J, Calderón-Sotelo P, Calle Jara DA, Camuglia JE, Cano MR, Capra M, Cassoux N, Castela G, Castillo L, Català-Mora J, Chantada GL, Chaudhry S, Chaugule SS, Chauhan A, Chawla B, Chernodrinska VS, Chiwanga FS, Chuluunbat T, Cieslik K, Cockcroft RL, Comsa C, Correa ZM, Correa Llano MG, Corson TW, Cowan-Lyn KE, Csóka M, Cui X, Da Gama IV, Dangboon W, Das A, Das S, Davanzo JM, Davidson A, De Potter P, Delgado KQ, Demirci H, Desjardins L, Diaz Coronado RY, Dimaras H, Dodgshun AJ, Donaldson C, Donato Macedo CR, Dragomir MD, Du Y, Du Bruyn M, Edison KS, Eka Sutyawan IW, El Kettani A, Elbahi AM, Elder JE, Elgalaly D, Elhaddad AM, Elhassan MMA, Elzembely MM, Essuman VA, Evina TGA, Fadoo Z, Fandiño AC, Faranoush M, Fasina O, Fernández DDPG, Fernández-Teijeiro A, Foster A, Frenkel S, Fu LD, Fuentes-Alabi SL, Gallie BL, Gandiwa M, Garcia JL, García Aldana D, Gassant PY, Geel JA, Ghassemi F, Girón AV, Gizachew Z, Goenz MA, Gold AS, Goldberg-Lavid M, Gole GA, Gomel N, Gonzalez E, Gonzalez Perez G, González-Rodríguez L, Garcia Pacheco HN, Graells J, Green L, Gregersen PA, Grigorovski NDAK, Guedenon KM, Gunasekera DS, Gündüz AK, Gupta H, Gupta S, Hadjistilianou T, Hamel P, Hamid SA, Hamzah N, Hansen ED, Harbour JW, Hartnett ME, Hasanreisoglu M, Hassan S, Hassan S, Hederova S, Hernandez J, Hernandez LMC, Hessissen L, Hordofa DF, Huang LC, Hubbard GB, Hummlen M, Husakova K, Hussein Al-Janabi AN, Ida R, Ilic VR, Jairaj V, Jeeva I, Jenkinson H, Ji X, Jo DH, Johnson KP, Johnson WJ, Jones MM, Kabesha TBA, Kabore RL, Kaliki S, Kalinaki A, Kantar M, Kao LY, Kardava T, Kebudi R, Kepak T, Keren-Froim N, Khan ZJ, Khaqan HA, Khauv P, Kheir WJ, Khetan V, Khodabande A, Khotenashvili Z, Kim JW, Kim JH, Kiratli H, Kivelä TT, Klett A, Komba Palet JEK, Krivaitiene D, Kruger M, Kulvichit K, Kuntorini MW, Kyara A, Lachmann ES, Lam CPS, Lam GC, Larson SA, Latinovic S, Laurenti KD, Le BHA, Lecuona K, Leverant AA, Li C, Limbu B, Long QB, López JP, Lukamba RM, Lumbroso L, Luna-Fineman S, Lutfi D, Lysytsia L, Magrath GN, Mahajan A, Majeed AR, Maka E, Makan M, Makimbetov EK, Manda C, Martín Begue N, Mason L, Mason JO, Matende IO, Materin M, Mattosinho CCDS, Matua M, Mayet I, Mbumba FB, McKenzie JD, Medina-Sanson A, Mehrvar A, Mengesha AA, Menon V, Mercado GJVD, Mets MB, Midena E, Mishra DKC, Mndeme FG, Mohamedani AA, Mohammad MT, Moll AC, Montero MM, Morales RA, Moreira C, Mruthyunjaya P, Msina MS, Msukwa G, Mudaliar SS, Muma KI, Munier FL, Murgoi G, Murray TG, Musa KO, Mushtaq A, Mustak H, Muyen OM, Naidu G, Nair AG, Naumenko L, Ndoye Roth PA, Nency YM, Neroev V, Ngo H, Nieves RM, Nikitovic M, Nkanga ED, Nkumbe H, Nuruddin M, Nyaywa M, Obono-Obiang G, Oguego NC, Olechowski A, Oliver SCN, Osei-Bonsu P, Ossandon D, Paez-Escamilla MA, Pagarra H, Painter SL, Paintsil V, Paiva L, Pal BP, Palanivelu MS, Papyan R, Parrozzani R, Parulekar M, Pascual Morales CR, Paton KE, Pawinska-Wasikowska K, Pe’er J, Peña A, Peric S, Pham CTM, Philbert R, Plager DA, Pochop P, Polania RA, Polyakov VG, Pompe MT, Pons JJ, Prat D, Prom V, Purwanto I, Qadir AO, Qayyum S, Qian J, Rahman A, Rahman S, Rahmat J, Rajkarnikar P, Ramanjulu R, Ramasubramanian A, Ramirez-Ortiz MA, Raobela L, Rashid R, Reddy MA, Reich E, Renner LA, Reynders D, Ribadu D, Riheia MM, Ritter-Sovinz P, Rojanaporn D, Romero L, Roy SR, Saab RH, Saakyan S, Sabhan AH, Sagoo MS, Said AMA, Saiju R, Salas B, San Román Pacheco S, Sánchez GL, Sayalith P, Scanlan TA, Schefler AC, Schoeman J, Sedaghat A, Seregard S, Seth R, Shah AS, Shakoor SA, Sharma MK, Sherief ST, Shetye NG, Shields CL, Siddiqui SN, Sidi Cheikh S, Silva S, Singh AD, Singh N, Singh U, Singha P, Sitorus RS, Skalet AH, Soebagjo HD, Sorochynska T, Ssali G, Stacey AW, Staffieri SE, Stahl ED, Stathopoulos C, Stirn Kranjc B, Stones DK, Strahlendorf C, Suarez MEC, Sultana S, Sun X, Sundy M, Superstein R, Supriyadi E, Surukrattanaskul S, Suzuki S, Svojgr K, Sylla F, Tamamyan G, Tan D, Tandili A, Tarrillo Leiva FF, Tashvighi M, Tateshi B, Tehuteru ES, Teixeira LF, Teh KH, Theophile T, Toledano H, Trang DL, Traoré F, Trichaiyaporn S, Tuncer S, Tyau-Tyau H, Umar AB, Unal E, Uner OE, Urbak SF, Ushakova TL, Usmanov RH, Valeina S, van Hoefen Wijsard M, Varadisai A, Vasquez L, Vaughan LO, Veleva-Krasteva NV, Verma N, Victor AA, Viksnins M, Villacís Chafla EG, Vishnevskia-Dai V, Vora T, Wachtel AE, Wackernagel W, Waddell K, Wade PD, Wali AH, Wang YZ, Weiss A, Wilson MW, Wime ADC, Wiwatwongwana A, Wiwatwongwana D, Wolley Dod C, Wongwai P, Xiang D, Xiao Y, Yam JC, Yang H, Yanga JM, Yaqub MA, Yarovaya VA, Yarovoy AA, Ye H, Yousef YA, Yuliawati P, Zapata López AM, Zein E, Zhang C, Zhang Y, Zhao J, Zheng X, Zhilyaeva K, Zia N, Ziko OAO, Zondervan M, Bowman R.. Global Retinoblastoma Presentation and Analysis by National Income Level. JAMA Oncol. 2020;6(5):685-695. doi:10.1001/jamaoncol.2019.6716. PMID:32105305; PMCID:PMC7047856.
Copia il testo dell'articolo e incollalo nell'assistente IA che preferisci.
Articolo copiato negli appunti
Apri un assistente IA qui sotto e incolla il testo copiato nella chat.