Retinoblastoma, bebek ve küçük çocukların retinasında oluşan kötü huylu bir tümördür. Olgunlaşmamış retina hücrelerinin kanserleşip çoğalarak kitle oluşturması durumudur ve 13. kromozomun uzun kolunda (13q14.2) yer alan RB1 genindeki mutasyonun neden olduğu tek gen hastalığı olarak kabul edilir. Cinsiyet farkı yoktur ve vakaların %95’i 5 yaşına kadar teşhis edilir.
Görülme sıklığı 15.000-23.000 doğumda 1’dir ve Japonya’da yılda 70-80 kişi hastalığa yakalanır. Tek taraflı ve çift taraflı olma oranı 3:2’dir ve teşhis zamanı tek taraflıda ortalama 21 ay, çift taraflıda ortalama 8 aydır; çift taraflı vakalar daha erken teşhis edilme eğilimindedir. Gelişmiş ülkelerde göz içine sınırlı evrede 5 yıllık sağkalım oranı %95’in üzerinde olup iyidir.
Retinoblastom, gen mutasyonu tipine göre iki ana gruba ayrılır.
Sınıflandırma
Mutasyon tipi
Hastalık özellikleri
Genetik risk
Kalıtsal (germline mutasyon)
Eşey hücrelerinde RB1 mutasyonu
Genellikle iki taraflı ve çoklu tümör
Çocuklara %50 oranında geçer
Kalıtsal olmayan (somatik mutasyon)
Bir retina hücresinde somatik mutasyon
Tek taraflı ve tek tümör
Sonraki nesle geçmez
Kalıtsal (germline mutasyon): Vücuttaki tüm hücrelerde ilk mutasyon bulunur. İkinci mutasyon oluştuğunda kanser gelişir. İki taraflı ve çoklu tümör görülme eğilimi yüksektir ve çocukların %50’sine geçer. Osteosarkom gibi ikincil kanser riski (20 yılda %15.7) vardır.
Kalıtsal olmayan (somatik mutasyon): Retinadaki bir hücrede RB1 geninin her iki kopyasında da mutasyon oluşması durumudur. Tek taraflı ve tek tümör şeklinde görülür ve sonraki nesle geçmez.
Ancak, tek taraflı vakaların bir kısmı da germ hattı RB1 mutasyonları içerir. Bu nedenle, tek taraflı olması kalıtsallığı dışlamaz; aile öyküsü, başlangıç yaşı ve tümör sayısı, genetik danışmanlık ve genetik değerlendirme temelinde yorumlanmalıdır. 1)
Evreleme, göz koruyucu tedavi stratejisiyle doğrudan ilişkilidir.
Evre
Lezyonun durumu
Göz korunma oranı tahmini
T1 (Erken intraoküler lezyon)
Göz içine sınırlı, ilerleme yok
%90’ın üzerinde
T2 (İlerlemiş intraoküler lezyon)
Göz içi ilerleme
Yaklaşık %50
T3
Ekstraoküler invazyonlu ilerlemiş lezyon
Yaklaşık %10
QRetinoblastom kalıtsal mıdır?
A
Yaklaşık %40’ı kalıtsaldır (germ hattı RB1 mutasyonu) ve %50 olasılıkla çocuğa geçer. Geri kalan %60’ı kalıtsal değildir (sadece somatik mutasyon) ve sonraki nesillere geçme riski yoktur. Kalıtsal olgularda bilateral ve multifokal olma eğilimi vardır. Tanı anında genetik test yapılması ve genetik danışmanlık alınması önerilir.
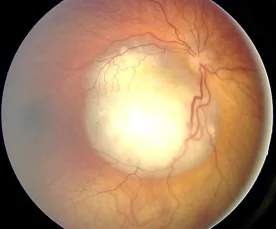
Aerts I, et al. Retinoblastoma. Orphanet J Rare Dis. 2006 Aug 25; 1:31. Figure 2. PMCID: PMC1586012. License: CC BY.
Retinoblastomun fundus görüntüsü, damardan zengin beyaz kabarık bir lezyon olarak izlenir ve lökokori (beyaz gözbebeği) neden olan tipik bulguyu gösterir. Bu görüntü, metnin “2. Ana belirtiler ve klinik bulgular” bölümünde ele alınan fundus bulgusu ve lökokoriye karşılık gelir.
Çoğu durumda tümör göz içinde büyür ve lökokori (beyaz gözbebeği) ile fark edilir. Makulada oluşursa, görme azlığı nedeniyle şaşılık ortaya çıkar ve bu şekilde teşhis edilebilir. Daha büyük çocuklarda görme azlığı fark edilmesi, küçük çocuklarda ise gözü ovuşturma hareketi ilk belirti olabilir.
İlk belirtiler
Lökokori (beyaz gözbebeği): En sık görülen ilk belirti. Tümör göz içinde büyür ve gözbebeği beyaz görünür.
Şaşılık: Makuladaki tümöre bağlı görme azlığından kaynaklanır. Görme azlığı olan göz dışa doğru kayar.
Görme azlığının fark edilmesi: Daha büyük çocuklarda görülür.
Gözü ovuşturma hareketi: Küçük çocuklarda görme azlığı olan gözde görülür.
İleri evre belirtileri
Kornea bulanıklığı ve göz içi basınç artışı: Tümörün lensi itmesi veya neovasküler glokom (NVG) nedeniyle göz içi basıncının artması sonucu oluşur.
Damardan zengin beyaz kabarık lezyonlar görülür ve kalsifikasyon eşlik ediyorsa kesin tanı kolaylaşır. Sıklıkla vitreus içine tümör hücrelerinin dökülmesi (vitreus tohumlanması) eşlik eder.
Kırmızı refle yöntemi, bebeklerde göz hastalıkları taramasının temelidir. Her iki göz bebeğinin boyutu eşit, parlak ve simetrik sarı-turuncu renkte ise normaldir. Refleks karanlık veya aşırı parlak ise ya da iki göz arasında fark varsa anormal kabul edilir ve ileri inceleme gerekir.
QBeyaz göz bebeği her zaman retinoblastom anlamına mı gelir?
A
Beyaz göz bebeğinin nedenleri arasında retinoblastom dışında fetal damar kalıntıları (primitif vitreus hiperplazisi), prematüre retinopatisi, Coats hastalığı gibi birçok durum vardır. Ancak beyaz göz bebeği görüldüğünde acilen göz doktoruna başvurulmalı ve retinoblastom dışlanması en öncelikli konudur. Tanıdaki gecikme prognozu doğrudan etkilediğinden, şüphe halinde aynı gün sevk edilmesi önerilir.
kromozomun uzun kolunda (13q14.2) bulunan RB1 genindeki mutasyon hastalığın nedenidir. RB1 geni, hücre bölünmesinin kontrolünde önemli rol oynayan RB1 proteinini (retinoblastom proteini) üretir.
Her hücrede iki gen kopyası bulunur; sadece bir kopyadaki mutasyon hücre fonksiyonunu korur, ancak her iki kopyada mutasyon olması hücre bölünmesinin kontrolünü kaybetmesine ve malignleşmeye yol açar (iki aşamalı karsinogenez teorisi, Knudson hipotezi).
Kalıtsal (germline mutasyon): İlk vuruş germ hattı mutasyonudur (tüm hücrelerde bulunur). Bir somatik hücrede ikinci vuruş meydana geldiğinde kanser oluşur. Bilateral ve multipl tümörler görülme eğilimi yüksektir.
Kalıtsal olmayan (somatik mutasyon): Hem birinci hem ikinci vuruş aynı somatik hücrede meydana gelir. Tek taraflı ve tek tümör şeklinde ortaya çıkar.
Kalıtsal vakalarda ikincil kanser riskine dikkat edilmelidir. Osteosarkom tipik bir örnektir ve kalıtsal vakalarda 20 yıl içinde %15.7 oranında görülür. İkincil kanserler genellikle 10’lu yaşlardan sonra ortaya çıkar.
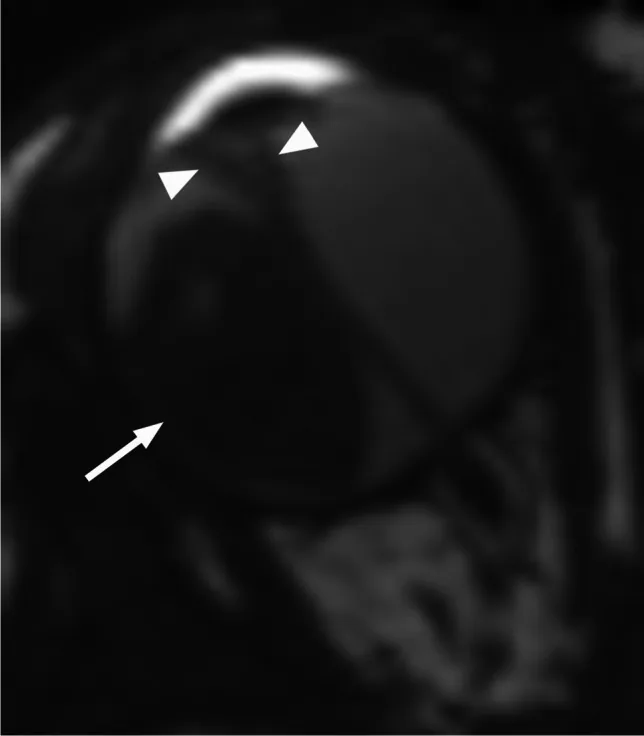
Rumboldt Z, Dodig D, Galluzzi P, et al. Retinoblastoma and beyond: pediatric orbital mass lesions. Neuroradiology. 2025;67(2):469-492. Figure 2. PMID: 39729290; PMCID: PMC11893699; DOI: 10.1007/s00234-024-03517-6. License: CC BY.
Aksiyel T2 ağırlıklı MRG’de ekzofitik retinoblastom (ok) ve V şeklinde sekonder retina dekolmanı (ok başı) görülmektedir. Bu görüntü, metnin “4. Tanı ve Test Yöntemleri” bölümünde tartışılan ekzofitik ve endofitik büyüme paternleri ve MRG bulgularına karşılık gelmektedir.
İç göz tümörlerinden biyopsi yapılmaz. Göz içi lezyonlar şeffaf doku aracılığıyla doğrudan gözlemlenebilir ve klinik tanının doğruluğu yüksektir. Ayrıca, iç göz tümörü biyopsisi tümör hücrelerinin göz dışına yayılmasına ve metastaz riskine yol açabilir. Göz koruyucu tedavi uygulanacaksa tedavi klinik tanıya dayanarak başlatılır.
Astrositik hamartom: Tümör damarlarının varlığı, OCT’de yerleşim ve büyüme varlığı ile ayırt edilir
Oküler sistiserkoz: Nadir olmakla birlikte Rb’yi taklit edebilir. Beyaz pupilla nedeniyle Rb şüphesiyle enükleasyon yapılan 4 yaşında bir erkek çocukta patolojik tanının sistiserkoz olduğu bildirilmiştir3)
Ailesel Rb’li çocukların taranması için AAOOP 2018 önerileri uluslararası alanda yaygın olarak kullanılmaktadır1).
Risk
Tarama takvimi
Bitiş zamanı
Yüksek (>%7.5)
Doğum-8 hafta: 2-4 haftada bir → 8-12 hafta: ayda bir → 1-2 yaş: 2 ayda bir → 2-3 yaş: 3 ayda bir → 3-4 yaş: 4 ayda bir → 4-7 yaş: 6 ayda bir
7 yaş (RB1 mutasyonu taşıyıcıları ömür boyu)
Orta (%1-7.5)
Doğum-3 ay: ayda bir → kademeli azaltma
7 yaş
Düşük (<%1)
Doğumdan 3 aya kadar: ayda 1 kez → kademeli azaltma
7 yaş
Hollanda ulusal verilerinin retrospektif kohort çalışmasında (1991-2019, 332 kişiden 38’i ailesel Rb) tam tarama alan 28 kişinin tamamına 1 yaşından önce (ortanca 18 gün) tanı konulurken, eksik tarama alan 10 kişide tanı ortanca 420 gün (59 gün-4,8 yıl) ile önemli ölçüde gecikmiştir2). Ayrıca düşük risk grubunda (<%3) tarama bitiş yaşının 2’ye düşürülmesi için protokol revizyonu önerilmiştir2).
Ailesel vakalarda, doğumdan hemen sonra başlayan fundus taramasının devamı doğrudan prognozla ilişkilidir. Klasik kayıt çalışmalarında bile, aile içi vakaların tanı zamanı tarama sıklığıyla yakından ilişkilidir ve günümüzde RB1 mutasyon varlığı/yokluğu ile birleştirilerek bitiş zamanının bireyselleştirilmesi yönünde ilerlenmektedir.1, 2)
QAilede retinoblastoma öyküsü varsa, çocuğun taraması ne kadar süre gerekir?
A
AAOOP önerisi 7 yaşına kadar düzenli fundus muayenesidir. Tam tarama yapıldığında, neredeyse tüm vakalar 1 yaşından önce teşhis edilir. Genetik test RB1 mutasyon riskini dışlarsa tarama erken sonlandırılabilir. RB1 mutasyon taşıyıcıları için 7 yaşından sonra 1-2 yılda bir düzensiz takip önerilir.
Görme fonksiyonunun korunabileceği erken intraoküler evrelerde, göz koruyucu tedavi aktif olarak uygulanır. İleri intraoküler evrelerde görme fonksiyonu genellikle beklenmez, ancak aile isterse koruyucu tedavi düşünülür. Tedavi yüksek uzmanlık gerektirir ve erken dönemde uzman merkeze sevk önemlidir.
Çapı yaklaşık 3 mm’ye kadar olan tümörler hedeftir. Kızılötesi lazerin doğrudan ışınlanmasıyla yaklaşık %90 lokal kontrol sağlanabilir. Makulada tümör varsa, geri dönüşümsüz görme kaybını önlemek için öncelikle sistemik kemoterapi önerilir.
② Kriyoterapi (dondurma)
Ekvator ve çevresinde yaklaşık 3 mm’lik tümörler tedavi hedefidir. Dondurma-çözme işleminin üç kez tekrarlandığı üçlü dondurma-çözme (triple freeze-thaw) yöntemi yaygındır ve lazer gibi yaklaşık %90 lokal kontrol sağlar.
③ Brakiterapi (lokal radyoterapi)
Bu yöntem, kalınlığı ≤5 mm ve çapı ≤15 mm olan, optik diskten uzak lokalize tümörler için uygundur. Japonya ve Avrupa’da ¹⁰⁶Ru (Rutenyum-106, beta kaynağı), Kuzey Amerika’da ise ¹²⁵I kaynağı kullanılır. Tedavide, tümöre karşılık gelen sklera yüzeyine geçici olarak bir radyoaktif kaynak dikilir. Bu işlem özel bir tedavi odası gerektirir ve sınırlı sayıda merkezde yapılır. Lokal kontrol oranı %80-90’dır.
④ Sistemik kemoterapi (VEC rejimi)
Bu tedavi, ilerlemiş intraoküler tümörlerde ilk seçenek olarak uygulanır. Üçlü ilaç kombinasyonu yaygın olarak kullanılır, ancak tek başına tedavi ile kür oranı %10’un altındadır. Tümör küçüldükten sonra lokal tedaviler (lazer, kriyoterapi, brakiterapi) ile pekiştirme yapılır.
İlaç
Doz (vücut yüzey alanına göre)
Doz (≤36 ay için kiloya göre)
Uygulama şeması
Vinkristin (Oncovin®)
1.5 mg/m²
0.05 mg/kg
1. gün
Karboplatin (Paraplatin®)
560 mg/m²
18.6 mg/kg
1. gün
Etoposid (Vepesid®)
150 mg/m²
5 mg/kg
gün 1, 2
Her 3-4 haftada bir, 2-6 kez tekrarlanır (tümü intravenöz infüzyon).
Kateter kullanılarak oftalmik artere doğrudan ilaç (melfalan; Alkeran® enjeksiyon çözeltisi) uygulanır. Gözün lokal bölgesine daha fazla ilaç verilerek sistemik doz azaltılır ve kemik iliği baskılanması gibi yan etkiler hafifletilir. Sigorta kapsamı dışındadır ancak 20’den fazla ülkede araştırma tedavisi olarak uygulanmaktadır.
⑥ İntravitreal Enjeksiyon
Vitreus tohumlanmasında sistemik kemoterapi ve arteriyel infüzyonun etkisi sınırlı olduğundan, melfalan (Alkeran® enjeksiyon çözeltisi) intravitreal enjeksiyonu ek olarak kullanılır. Sigorta kapsamı dışında araştırma tedavisidir.
⑦ Harici Radyoterapi
40-46 Gy x-ışını fraksiyone olarak uygulanır. 1990’lara kadar göz koruyucu tedavinin temelini oluşturuyordu ancak orbital kemik deformitesi ve ikincil kanser artışı belirgin hale geldiğinden, günümüzde yalnızca diğer tedavilerle kontrol edilemeyen durumlarda sınırlı olarak yapılmaktadır.
Optik sinir pozitif cerrahi sınırı ve ekstraskleral invazyon, ameliyat sonrası tedavi için mutlak endikasyonlardır ve sistemik kemoterapi ile radyoterapi uygulanır. Belirgin koroid invazyonu veya lamina kribrozayı aşan optik sinir invazyonu, metastaz için göreceli risk faktörleri olarak değerlendirilir.
Göz korunmasının mümkün olduğu durumlar
T1 (intraoküler erken lezyon): Korunma oranı %90’ın üzerinde
Tümör çapı 3 mm veya daha küçük: Lazer tedavisi veya kriyokoagülasyon ilk seçenek
Tümör kalınlığı 5 mm veya daha küçük: Brakiterapi uygundur
Sistemik kemoterapi → lokal tedavi: İleri vakalarda bile küçülme sonrası korunma denenir
Gözün alınmasını gerektiren durumlar
T3 (ekstraoküler invazyon): Korunma oranı yaklaşık %10
Ön kamara veya iris invazyonu: Ekstraoküler yayılma riski
Görme fonksiyonunun düzelmesi beklenmediğinde: Yaşam prognozuna öncelik verilir
QGözü koruma olasılığı var mı?
A
Erken evre intraoküler lezyonlarda (T1) %90’ın üzerinde göz korunabilir. Sistemik kemoterapi (VEC rejimi) ile tümör küçültülür, ardından lazer, kriyoterapi ve brakiterapi ile pekiştirme yapılır. İleri evrelerde (T3) göz koruma oranı yaklaşık %10’dur ve enükleasyon gerekebilir. Tedavi seçimi, evre ve görme potansiyeline göre uzman hekim tarafından belirlenir.
kromozomun uzun kolunda (13q14.2) yer alan RB1 geni, hücre bölünmesinin kontrolünde önemli rol oynayan RB1 proteinini (pRb) kodlar. pRb, E2F transkripsiyon faktörüne bağlanarak hücre döngüsünün G1/S geçişini inhibe eder ve böylece hücre çoğalmasını kontrol eden bir tümör baskılayıcı proteindir.
Knudson tarafından öne sürülen iki vuruş teorisine göre, bir hücredeki RB1 geninin her iki alelinin de inaktive olması malign transformasyona yol açar.
Kalıtsal: İlk vuruş (germ hattı mutasyonu) tüm hücrelerde bulunur. Retina hücrelerinden birinde ikinci vuruş (somatik mutasyon, LOH vb.) meydana geldiğinde kanser oluşur. Bu nedenle bilateral ve multifokal tümörler sık görülür ve nispeten erken teşhis edilir.
Kalıtsal olmayan: Hem birinci hem de ikinci vuruş aynı retina hücresinde somatik mutasyon olarak ortaya çıkar. Her iki vuruşun aynı hücrede tesadüfen bir araya gelme olasılığı düşük olduğundan, tümörler genellikle tek taraflı ve soliterdir ve tanı kalıtsal forma göre daha geç konur.
Kalıtsal olgularda, vücuttaki tüm hücrelerde RB1’in ilk vuruşu bulunur. Retina dışındaki hücrelerde (kemik, yumuşak doku vb.) ikinci vuruş meydana gelirse ikincil primer malign tümör gelişir. En sık osteosarkom görülür ve genellikle 10’lu yaşlardan sonra ortaya çıkar. Radyoterapi alan kalıtsal olgularda ikincil kanser riski daha da arttığından, günümüzde eksternal radyoterapi sınırlı olarak kullanılmaktadır.
IAC (intra-arteriyel kemoterapi), melfalanın oftalmik artere doğrudan enjeksiyonu ile sistemik toksisiteyi en aza indirirken göz içine yüksek konsantrasyonda ilaç ulaştırılmasını sağlar. Vitreus tohumlanması olan ilerlemiş vakalarda bile göz koruma şansını artırabilir. Halen büyük kohortlarda uzun dönem sonuçların değerlendirilmesi devam etmektedir.
Vitreus tohumlanmasına yönelik olarak intravitreal melfalan enjeksiyonu uygulanmaktadır, ancak uluslararası doz ve uygulama aralığı protokolünün standardizasyonu bir zorluktur. Birden fazla vaka serisinde yüksek tohumlanma kontrol oranları bildirilmiş olup, gelecekteki prospektif çalışmalarla yerinin belirlenmesi beklenmektedir.
Hollanda kohort çalışmasında, tam taramaya katılım ile erken tanı (ortanca 18 gün) arasında net bir korelasyon gösterilmiş ve taramanın kesilmesinin zararları nicel olarak belirlenmiştir 2). Ayrıca düşük riskli gruplarda tarama bitiş yaşının kısaltılması (2 yaş) gibi risk sınıflandırmasına dayalı protokol optimizasyonu ilerlemektedir 2). AAOOP önerilerinin dünya çapında yaygınlaştırılması ve bölgesel protokollerin standardizasyonu gelecekteki zorluklardır 1).
Nadir olmakla birlikte, konjenital beyin malformasyonları veya kromozomal anormallikler zemininde retinoblastomun eşlik ettiği vaka raporları mevcuttur. Dandy-Walker sendromlu bilateral retinoblastom raporunda, oküler semptomların yanı sıra nörogelişimsel arka planı da içeren tam sistemik değerlendirmenin önemi vurgulanmıştır. 4)
Yeni nesil dizilemenin (NGS) yaygınlaşmasıyla, germ hattı RB1 mutasyonlarının tespit duyarlılığı artmıştır. Sıvı biyopsi (dolaşımdaki tümör DNA’sı) kullanılarak izleme olasılığı da araştırılmakta olup, invaziv biyopsiden kaçınarak prognoz sınıflandırmasına uygulanması beklenmektedir.
Kalıtsal retinoblastomun uzun dönem sağ kalanları için ikincil kanser tarama protokolünün standardizasyonu gereklidir. Özellikle tüm vücut MRI ile çoklu organ taramasının sıklığı ve bitiş zamanı konusunda sürekli kohort çalışmalarıyla kanıt birikimi sağlanmaktadır.
Selektif oftalmik arter infüzyonu (IAC) ve intravitreal kemoterapi (IVitC), ilerlemiş vakalar ve vitreus tohumlanması olan vakalarda göz koruma oranlarını artıran tedaviler olarak uluslararası alanda yerleşmektedir. Konservatif tedavi derlemelerinde, metastaz riskini artırmadan göz korumayı genişleten stratejiler olarak konumlandırılmışlardır ve ülkelerin uygulama durumu ile uzman merkezlerde yoğunlaşma sonuçları etkilemektedir. 5)
Gelişmiş ülkelerde 5 yıllık sağkalım oranı %95’in üzerindeyken, Afrika ve Asya’daki düşük ve orta gelirli ülkelerde bu oranın %25-70 arasında kaldığı bildirilmektedir. Sistematik derlemeler ve ülkelerin gelir düzeyine göre yapılan analizler, sağkalım ve göz koruma oranlarındaki eşitsizliği tutarlı bir şekilde göstermekte olup, başlıca nedenler geç tanı, sağlık hizmetlerine yetersiz erişim ve uzman tesis eksikliği olarak belirtilmektedir. Kırmızı refleks yöntemini de içeren toplum temelli tarama programlarının yaygınlaştırılması uluslararası bir zorluktur. 6, 7)
Skalet AH, Gombos DS, Gallie BL, Kim JW, Shields CL, Marr BP, Plon SE, Chévez-Barrios P. Screening Children at Risk for Retinoblastoma: Consensus Report from the American Association of Ophthalmic Oncologists and Pathologists. Ophthalmology. 2018;125(3):453-458. doi:10.1016/j.ophtha.2017.09.001. PMID:29056300.
Badalova NA, van Hoefen Wijsard M, Dommering CJ, et al. At what age could screening for familial retinoblastoma be stopped? Revised Dutch retrospective population-based cohort study 1991-2019. Ophthalmology. 2024;131(10):1189-1196.
Jakati S, Vempuluru VS, Kaliki S. Intraocular Cysticercosis Masquerading as Cavitary Retinoblastoma. Ophthalmology. 2025;132(4):e68. doi:10.1016/j.ophtha.2024.05.017. PMID:38878044.
Lomi N, Das D, Chawla B, Parampalli Ravindra A. Retinoblastoma in Dandy-Walker Syndrome. Cureus. 2025;17(8):e89663. doi:10.7759/cureus.89663. PMID:40926918; PMCID:PMC12415497.
Francis L. Munier, Maja Beck-Popovic, Guillermo L. Chantada, David Cobrinik, Tero T. Kivelä, Dietmar Lohmann, Philippe Maeder, Annette C. Moll, et al. Conservative management of retinoblastoma: Challenging orthodoxy without compromising the state of metastatic grace. “Alive, with good vision and no comorbidity”. Progress in Retinal and Eye Research. 2019;73:100764. doi:10.1016/j.preteyeres.2019.05.005.
Wong ES, Choy RW, Zhang Y, et al. Global retinoblastoma survival and globe preservation: a systematic review and meta-analysis. Lancet Glob Health. 2022;10(3):e380-e389.
Global Retinoblastoma Study Group, Fabian ID, Abdallah E, Abdullahi SU, Abdulqader RA, Adamou Boubacar S, Ademola-Popoola DS, Adio A, Afshar AR, Aggarwal P, Aghaji AE, Ahmad A, Akib MNR, Al Harby L, Al Ani MH, Alakbarova A, Portabella SA, Al-Badri SAF, Alcasabas APA, Al-Dahmash SA, Alejos A, Alemany-Rubio E, Alfa Bio AI, Alfonso Carreras Y, Al-Haddad C, Al-Hussaini HHY, Ali AM, Alia DB, Al-Jadiry MF, Al-Jumaily U, Alkatan HM, All-Eriksson C, Al-Mafrachi AARM, Almeida AA, Alsawidi KM, Al-Shaheen AASM, Al-Shammary EH, Amiruddin PO, Antonino R, Astbury NJ, Atalay HT, Atchaneeyasakul LO, Atsiaya R, Attaseth T, Aung TH, Ayala S, Baizakova B, Balaguer J, Balayeva R, Balwierz W, Barranco H, Bascaran C, Beck Popovic M, Benavides R, Benmiloud S, Bennani Guebessi N, Berete RC, Berry JL, Bhaduri A, Bhat S, Biddulph SJ, Biewald EM, Bobrova N, Boehme M, Boldt HC, Bonanomi MTBC, Bornfeld N, Bouda GC, Bouguila H, Boumedane A, Brennan RC, Brichard BG, Buaboonnam J, Calderón-Sotelo P, Calle Jara DA, Camuglia JE, Cano MR, Capra M, Cassoux N, Castela G, Castillo L, Català-Mora J, Chantada GL, Chaudhry S, Chaugule SS, Chauhan A, Chawla B, Chernodrinska VS, Chiwanga FS, Chuluunbat T, Cieslik K, Cockcroft RL, Comsa C, Correa ZM, Correa Llano MG, Corson TW, Cowan-Lyn KE, Csóka M, Cui X, Da Gama IV, Dangboon W, Das A, Das S, Davanzo JM, Davidson A, De Potter P, Delgado KQ, Demirci H, Desjardins L, Diaz Coronado RY, Dimaras H, Dodgshun AJ, Donaldson C, Donato Macedo CR, Dragomir MD, Du Y, Du Bruyn M, Edison KS, Eka Sutyawan IW, El Kettani A, Elbahi AM, Elder JE, Elgalaly D, Elhaddad AM, Elhassan MMA, Elzembely MM, Essuman VA, Evina TGA, Fadoo Z, Fandiño AC, Faranoush M, Fasina O, Fernández DDPG, Fernández-Teijeiro A, Foster A, Frenkel S, Fu LD, Fuentes-Alabi SL, Gallie BL, Gandiwa M, Garcia JL, García Aldana D, Gassant PY, Geel JA, Ghassemi F, Girón AV, Gizachew Z, Goenz MA, Gold AS, Goldberg-Lavid M, Gole GA, Gomel N, Gonzalez E, Gonzalez Perez G, González-Rodríguez L, Garcia Pacheco HN, Graells J, Green L, Gregersen PA, Grigorovski NDAK, Guedenon KM, Gunasekera DS, Gündüz AK, Gupta H, Gupta S, Hadjistilianou T, Hamel P, Hamid SA, Hamzah N, Hansen ED, Harbour JW, Hartnett ME, Hasanreisoglu M, Hassan S, Hassan S, Hederova S, Hernandez J, Hernandez LMC, Hessissen L, Hordofa DF, Huang LC, Hubbard GB, Hummlen M, Husakova K, Hussein Al-Janabi AN, Ida R, Ilic VR, Jairaj V, Jeeva I, Jenkinson H, Ji X, Jo DH, Johnson KP, Johnson WJ, Jones MM, Kabesha TBA, Kabore RL, Kaliki S, Kalinaki A, Kantar M, Kao LY, Kardava T, Kebudi R, Kepak T, Keren-Froim N, Khan ZJ, Khaqan HA, Khauv P, Kheir WJ, Khetan V, Khodabande A, Khotenashvili Z, Kim JW, Kim JH, Kiratli H, Kivelä TT, Klett A, Komba Palet JEK, Krivaitiene D, Kruger M, Kulvichit K, Kuntorini MW, Kyara A, Lachmann ES, Lam CPS, Lam GC, Larson SA, Latinovic S, Laurenti KD, Le BHA, Lecuona K, Leverant AA, Li C, Limbu B, Long QB, López JP, Lukamba RM, Lumbroso L, Luna-Fineman S, Lutfi D, Lysytsia L, Magrath GN, Mahajan A, Majeed AR, Maka E, Makan M, Makimbetov EK, Manda C, Martín Begue N, Mason L, Mason JO, Matende IO, Materin M, Mattosinho CCDS, Matua M, Mayet I, Mbumba FB, McKenzie JD, Medina-Sanson A, Mehrvar A, Mengesha AA, Menon V, Mercado GJVD, Mets MB, Midena E, Mishra DKC, Mndeme FG, Mohamedani AA, Mohammad MT, Moll AC, Montero MM, Morales RA, Moreira C, Mruthyunjaya P, Msina MS, Msukwa G, Mudaliar SS, Muma KI, Munier FL, Murgoi G, Murray TG, Musa KO, Mushtaq A, Mustak H, Muyen OM, Naidu G, Nair AG, Naumenko L, Ndoye Roth PA, Nency YM, Neroev V, Ngo H, Nieves RM, Nikitovic M, Nkanga ED, Nkumbe H, Nuruddin M, Nyaywa M, Obono-Obiang G, Oguego NC, Olechowski A, Oliver SCN, Osei-Bonsu P, Ossandon D, Paez-Escamilla MA, Pagarra H, Painter SL, Paintsil V, Paiva L, Pal BP, Palanivelu MS, Papyan R, Parrozzani R, Parulekar M, Pascual Morales CR, Paton KE, Pawinska-Wasikowska K, Pe’er J, Peña A, Peric S, Pham CTM, Philbert R, Plager DA, Pochop P, Polania RA, Polyakov VG, Pompe MT, Pons JJ, Prat D, Prom V, Purwanto I, Qadir AO, Qayyum S, Qian J, Rahman A, Rahman S, Rahmat J, Rajkarnikar P, Ramanjulu R, Ramasubramanian A, Ramirez-Ortiz MA, Raobela L, Rashid R, Reddy MA, Reich E, Renner LA, Reynders D, Ribadu D, Riheia MM, Ritter-Sovinz P, Rojanaporn D, Romero L, Roy SR, Saab RH, Saakyan S, Sabhan AH, Sagoo MS, Said AMA, Saiju R, Salas B, San Román Pacheco S, Sánchez GL, Sayalith P, Scanlan TA, Schefler AC, Schoeman J, Sedaghat A, Seregard S, Seth R, Shah AS, Shakoor SA, Sharma MK, Sherief ST, Shetye NG, Shields CL, Siddiqui SN, Sidi Cheikh S, Silva S, Singh AD, Singh N, Singh U, Singha P, Sitorus RS, Skalet AH, Soebagjo HD, Sorochynska T, Ssali G, Stacey AW, Staffieri SE, Stahl ED, Stathopoulos C, Stirn Kranjc B, Stones DK, Strahlendorf C, Suarez MEC, Sultana S, Sun X, Sundy M, Superstein R, Supriyadi E, Surukrattanaskul S, Suzuki S, Svojgr K, Sylla F, Tamamyan G, Tan D, Tandili A, Tarrillo Leiva FF, Tashvighi M, Tateshi B, Tehuteru ES, Teixeira LF, Teh KH, Theophile T, Toledano H, Trang DL, Traoré F, Trichaiyaporn S, Tuncer S, Tyau-Tyau H, Umar AB, Unal E, Uner OE, Urbak SF, Ushakova TL, Usmanov RH, Valeina S, van Hoefen Wijsard M, Varadisai A, Vasquez L, Vaughan LO, Veleva-Krasteva NV, Verma N, Victor AA, Viksnins M, Villacís Chafla EG, Vishnevskia-Dai V, Vora T, Wachtel AE, Wackernagel W, Waddell K, Wade PD, Wali AH, Wang YZ, Weiss A, Wilson MW, Wime ADC, Wiwatwongwana A, Wiwatwongwana D, Wolley Dod C, Wongwai P, Xiang D, Xiao Y, Yam JC, Yang H, Yanga JM, Yaqub MA, Yarovaya VA, Yarovoy AA, Ye H, Yousef YA, Yuliawati P, Zapata López AM, Zein E, Zhang C, Zhang Y, Zhao J, Zheng X, Zhilyaeva K, Zia N, Ziko OAO, Zondervan M, Bowman R.. Global Retinoblastoma Presentation and Analysis by National Income Level. JAMA Oncol. 2020;6(5):685-695. doi:10.1001/jamaoncol.2019.6716. PMID:32105305; PMCID:PMC7047856.
Makale metnini kopyalayıp tercih ettiğiniz yapay zeka asistanına yapıştırabilirsiniz.
Makale panoya kopyalandı
Aşağıdaki yapay zeka asistanlarından birini açın ve kopyalanan metni sohbet kutusuna yapıştırın.