O retinoblastoma é um tumor maligno que surge na retina de lactentes e crianças pequenas. É uma condição na qual células imaturas da retina se tornam cancerosas e proliferam formando uma massa tumoral, sendo considerada uma doença monogênica causada por mutações no gene RB1 localizado no braço longo do cromossomo 13 (13q14.2). Não há diferença entre sexos, e 95% são diagnosticados antes dos 5 anos.
A frequência é de 1 a cada 15.000-23.000 nascimentos, com 70-80 casos por ano no Japão. A proporção entre unilateral e bilateral é de 3:2, com idade média ao diagnóstico de 21 meses para unilateral e 8 meses para bilateral, indicando diagnóstico mais precoce. A taxa de sobrevida em 5 anos em países desenvolvidos é superior a 95% para doença intraocular limitada.
O retinoblastoma é dividido em dois tipos principais de acordo com o tipo de mutação genética.
Classificação
Tipo de mutação
Características da doença
Risco genético
Hereditário (mutação germinativa)
Mutação RB1 na linhagem germinativa
Frequentemente bilateral e tumores múltiplos
Transmitido a 50% dos filhos
Não hereditário (mutação somática)
Mutação somática em uma célula da retina
Tumor unilateral e solitário
Sem transmissão para a próxima geração
Hereditário (mutação germinativa): Estado em que a primeira mutação está presente em todas as células do corpo. Quando ocorre a segunda mutação, desenvolve-se câncer. Tende a produzir tumores bilaterais e múltiplos, sendo transmitido a 1 em cada 2 filhos (50%). Há risco de cânceres secundários como osteossarcoma (15,7% em 20 anos).
Não hereditário (mutação somática): Quando ocorrem mutações em ambos os loci do gene RB1 em uma única célula da retina. Apresenta-se como tumor unilateral e solitário, sem risco de transmissão para a próxima geração.
No entanto, mesmo em casos unilaterais, alguns incluem mutações germinativas do RB1. Não se deve negar a hereditariedade apenas por ser unilateral; é necessário interpretar a história familiar, idade de início e número de tumores com base em aconselhamento genético e avaliação genética. 1)
Classificação de Estadiamento e Taxa de Preservação do Olho
A classificação de estadiamento está diretamente ligada à estratégia de tratamento para preservação do olho.
Estádio
Estado da Lesão
Taxa de Preservação do Olho (Estimativa)
T1 (Estádio inicial intraocular)
Localizado intraocularmente, sem progressão
>90%
T2 (Estádio avançado intraocular)
Progressão intraocular
Cerca de 50%
T3
Lesão avançada com invasão extraocular
Cerca de 10%
QO retinoblastoma é hereditário?
A
Cerca de 40% são hereditários (mutação RB1 na linhagem germinativa) e são transmitidos aos filhos com 50% de probabilidade. Os outros 60% são não hereditários (apenas mutação somática) e não há risco hereditário para as próximas gerações. Nos casos hereditários, há tendência a bilateralidade e multifocalidade. Recomenda-se realizar teste genético no momento do diagnóstico e receber aconselhamento genético.
Aerts I, et al. Retinoblastoma. Orphanet J Rare Dis. 2006 Aug 25; 1:31. Figure 2. PMCID: PMC1586012. License: CC BY.
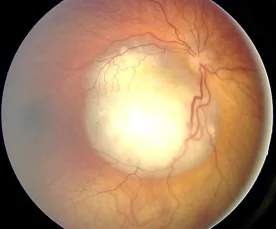
Imagem de fundo de olho do retinoblastoma observada como lesão elevada branca e vascularizada, mostrando achado típico que causa leucocoria. Corresponde à seção “2. Principais sintomas e achados clínicos” que aborda achados de fundo de olho e leucocoria.
Na maioria dos casos, o tumor cresce dentro do olho e é descoberto quando apresenta leucocoria. Quando ocorre na mácula, pode causar baixa visão e estrabismo, levando à descoberta. Em crianças mais velhas, a percepção de diminuição da visão pode ser o sintoma inicial; em crianças pequenas, o hábito de esfregar o olho com baixa visão pode ser o sintoma inicial.
Sintomas iniciais
Leucocoria: Sintoma inicial mais comum. O tumor cresce dentro do olho e a pupila parece branca.
Estrabismo: Causado pela baixa visão devido a tumor na mácula. O olho com baixa visão tende a desviar para fora.
Percepção de diminuição da visão: Observada em crianças mais velhas.
Hábito de esfregar o olho: Observado em crianças pequenas no olho com baixa visão.
Sintomas em estágio avançado
Opacidade corneana e aumento da pressão intraocular: Ocorrem devido à compressão do cristalino pelo tumor ou ao aumento da pressão intraocular por glaucoma neovascular (NVG).
Hiperemia conjuntival e edema palpebral: Observados como sinais inflamatórios.
Dor: Surge devido ao aumento da pressão intraocular ou necrose tumoral.
Proptose: Observada quando ocorre invasão extraocular.
Observa-se uma lesão elevada e esbranquiçada, rica em vasos sanguíneos; se houver calcificação, o diagnóstico é facilitado. Frequentemente acompanhada de disseminação vítrea (células tumorais se desprendem e se espalham no vítreo).
O método do reflexo vermelho é fundamental na triagem de doenças oculares em lactentes. É normal se as pupilas forem de tamanho igual e o reflexo for amarelo-alaranjado brilhante e simétrico. Se o reflexo for escuro ou muito claro, ou se houver diferença entre os olhos, é considerado anormal e requer investigação.
QLeucocoria significa necessariamente retinoblastoma?
A causa é uma mutação no gene RB1 localizado no braço longo do cromossomo 13 (13q14.2). O gene RB1 produz a proteína RB1 (proteína do retinoblastoma), que desempenha um papel importante no controle da divisão celular.
Em uma célula, existem dois loci gênicos; uma mutação em apenas um locus ainda mantém a função celular, mas quando ambos os loci sofrem mutação, a célula perde o controle da divisão e se torna maligna (teoria dos dois eventos, hipótese de Knudson).
Hereditário (mutação germinativa): O primeiro evento é uma mutação na linhagem germinativa (presente em todas as células). Quando o segundo evento ocorre em uma célula somática, ocorre a carcinogênese. Tendência a tumores bilaterais e múltiplos.
Não hereditário (mutação somática): Tanto o primeiro quanto o segundo evento ocorrem em uma única célula somática. Apresenta-se como tumor unilateral e solitário.
O histórico familiar é o maior fator de risco. A seguir, a definição de risco de acordo com as recomendações da AAOOP (Academia Americana de Oncologia e Patologia Oftálmica)1).
Classificação de Risco
Definição
Valor de Risco
Alto
Pai/mãe com Rb bilateral, ou portador de mutação germinativa RB1 em parentes de primeiro ou segundo grau
Em casos hereditários, é necessário estar atento ao risco de câncer secundário. Osteossarcoma é um exemplo típico, ocorrendo em 15,7% dos casos hereditários em 20 anos. O câncer secundário geralmente se desenvolve após a adolescência.
Rumboldt Z, Dodig D, Galluzzi P, et al. Retinoblastoma and beyond: pediatric orbital mass lesions. Neuroradiology. 2025;67(2):469-492. Figure 2. PMID: 39729290; PMCID: PMC11893699; DOI: 10.1007/s00234-024-03517-6. License: CC BY.
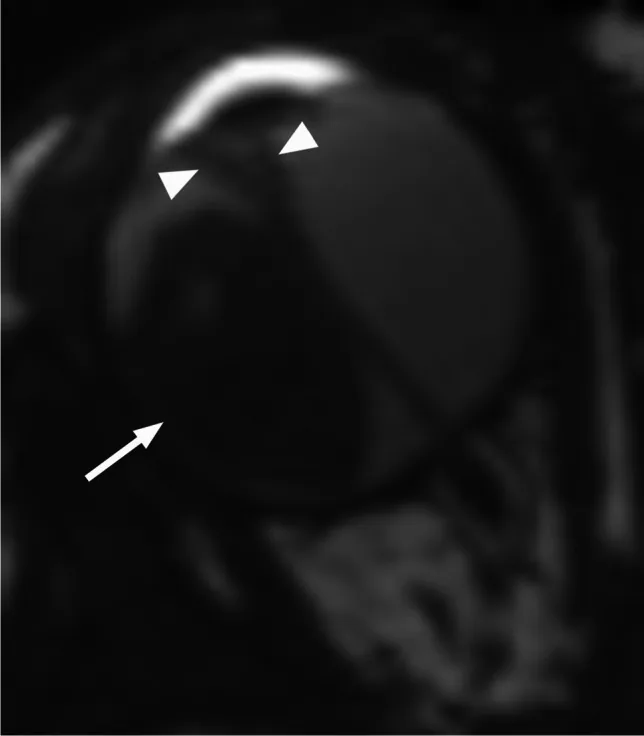
RM axial ponderada em T2 mostra retinoblastoma de crescimento exofítico (seta) com descolamento de retina secundário em forma de V (ponta de seta). Isso corresponde aos padrões de crescimento exofítico e endofítico e aos achados de RM discutidos na seção “4. Diagnóstico e Métodos de Exame”.
A biópsia de tumor intraocular não é realizada. As lesões intraoculares podem ser observadas diretamente através de tecidos transparentes, e a precisão do diagnóstico clínico é alta. Além disso, a biópsia de tumor intraocular pode causar disseminação de células tumorais para fora do globo ocular, resultando em risco inevitável de metástase. Quando a terapia de preservação ocular é realizada, o tratamento é iniciado com base no diagnóstico clínico.
Exame de fundo de olho (principal): Lesão elevada branca e vascularizada + calcificação para diagnóstico clínico definitivo
Ultrassonografia: Confirmação de tumor sólido e calcificação. Observação: calcificação é rara acima de 5 anos
RM: Sinal intermediário em T1, sinal levemente hipointenso em T2, com realce pelo contraste. Essencial para avaliar invasão do nervo óptico e invasão extraocular
RM de crânio: Cerca de 3% dos casos bilaterais desenvolvem retinoblastoma trilateral (tumor da glândula pineal), portanto a triagem é obrigatória
TC: Excelente para demonstrar calcificação, mas com exposição à radiação. Se a RM estiver disponível, a TC é complementar
O mais importante é diferenciar as doenças que causam leucocoria (reflexo pupilar branco).
Persistência do vítreo primário (hiperplasia vítrea primária persistente): Ultrassonografia para confirmar presença ou ausência de tumor parenquimatoso
Retinopatia da prematuridade: História de prematuridade e baixo peso ao nascer são pistas para o diagnóstico diferencial
Doença de Coats: Acúmulo de exsudato amarelo-esbranquiçado sob a retina. Padrão vascular tumoral diferente
Hamartoma astrocítico: Diferenciação baseada na presença ou ausência de vasos tumorais, localização na OCT e presença ou ausência de crescimento
Cisticercose ocular (cysticercosis): Raramente pode mimetizar Rb. Há relato de caso de um menino de 4 anos com pupila branca submetido a enucleação por suspeita de Rb, cujo diagnóstico patológico foi cisticercose3)
Em relação à triagem de crianças com história familiar de Rb familiar, a recomendação AAOOP 2018 é amplamente referenciada internacionalmente1).
Risco
Cronograma de triagem
Momento de término
Alto (>7,5%)
Nascimento–8 semanas: a cada 2–4 semanas → 8–12 semanas: 1 vez/mês → 1–2 anos: a cada 2 meses → 2–3 anos: a cada 3 meses → 3–4 anos: a cada 4 meses → 4–7 anos: a cada 6 meses
7 anos (portadores de mutação RB1 por toda a vida)
Intermediário (1–7,5%)
Nascimento–3 meses: 1 vez/mês → redução gradual
7 anos
Baixo (<1%)
Nascimento a 3 meses: 1 vez/mês → redução gradual
7 anos
Em um estudo de coorte retrospectivo holandês (1991–2019, 38 de 332 pacientes com Rb familiar), todos os 28 que realizaram triagem completa foram diagnosticados antes de 1 ano de idade (mediana 18 dias), enquanto os 10 com triagem incompleta tiveram diagnóstico com mediana de 420 dias (59 dias–4,8 anos)2). Também foi proposta uma revisão do protocolo para reduzir a idade de término da triagem para 2 anos no grupo de baixo risco (<3%)2).
Em casos familiares, a continuidade da triagem de fundo de olho desde o nascimento está diretamente ligada ao prognóstico. Mesmo em estudos de registro clássicos, o momento do diagnóstico intrafamiliar está intimamente relacionado à frequência da triagem, e atualmente há uma tendência à individualização do término da triagem com base na presença ou ausência de mutação RB1.1, 2)
QSe houver histórico familiar de retinoblastoma, até quando a criança precisa ser rastreada?
A
A AAOOP recomenda exame de fundo de olho regular até os 7 anos de idade. Se a triagem completa for realizada, a maioria é diagnosticada antes de 1 ano. Se o risco de mutação RB1 for descartado por teste genético, a triagem pode ser interrompida mais cedo. Portadores de mutação RB1 são recomendados a realizar acompanhamento irregular a cada 1–2 anos mesmo após os 7 anos.
Em lesões intraoculares iniciais com expectativa de boa função visual, o tratamento de preservação ocular é realizado ativamente. Em estágios intraoculares avançados, a função visual muitas vezes não é esperada, mas o tratamento de preservação pode ser considerado se a família desejar. O tratamento requer alto nível de especialização, sendo importante o encaminhamento precoce a um centro especializado.
Tumores com até cerca de 3 mm de diâmetro são alvo. O controle local de cerca de 90% pode ser alcançado por irradiação direta com laser infravermelho. Se o tumor estiver na mácula, recomenda-se quimioterapia sistêmica primeiro para evitar dano visual irreversível.
② Criocoagulação
Tumores de cerca de 3 mm na região periférica ao equador são alvo. O método de congelamento e descongelamento triplo (triple freeze-thaw), repetido 3 vezes, é comum, e o controle local de cerca de 90% é obtido, similar ao laser.
③ Braquiterapia
Indicada para tumores limitados com espessura ≤5 mm e diâmetro ≤15 mm, distantes do disco óptico. No Japão e Europa, utiliza-se fonte de ¹⁰⁶Ru (rutênio-106, emissor β); na América do Norte, fonte de ¹²⁵I. A fonte é suturada temporariamente na superfície escleral correspondente ao tumor. Requer sala de tratamento especial, limitando as instalações. O controle local é de 80-90%.
④ Quimioterapia sistêmica (VEC)
Realizada como primeira linha para tumores intraoculares em estágio avançado. A quimioterapia combinada com três agentes é amplamente utilizada, mas a cura com tratamento isolado é inferior a 10%. Após a redução tumoral, realiza-se tratamento local (laser, criocoagulação, braquiterapia) para consolidação.
Medicamento
Dose (com base na área de superfície corporal)
Dose (com base no peso para ≤36 meses)
Esquema de administração
Vincristina (Oncovin®)
1,5 mg/m²
0,05 mg/kg
Dia 1
Carboplatina (Paraplatin®)
560 mg/m²
18,6 mg/kg
Dia 1
Etoposídeo (Vepesid®)
150 mg/m²
5 mg/kg
dia 1, 2
Repetir 2 a 6 ciclos a cada 3-4 semanas (todos por infusão intravenosa).
O medicamento (melfalano; solução injetável Alkeran®) é administrado diretamente na artéria oftálmica por meio de um cateter. Ao administrar uma dose maior localmente no olho e uma dose menor sistemicamente, os efeitos colaterais como supressão da medula óssea podem ser reduzidos. Este é um tratamento experimental não coberto pelo seguro, mas realizado em mais de 20 países no mundo.
⑥ Injeção intravítrea
Para semeadura vítrea, o efeito da quimioterapia sistêmica e da injeção arterial é limitado, portanto, a injeção intravítrea de melfalano (solução injetável Alkeran®) é usada em conjunto. Este é um tratamento experimental não coberto pelo seguro.
⑦ Radioterapia externa
Raios-X são administrados em doses fracionadas de 40 a 46 Gy. Foi o pilar do tratamento de preservação ocular até a década de 1990, mas após a deformidade orbital e o aumento de cânceres secundários se tornarem evidentes, agora é usado apenas quando não pode ser controlado por outros tratamentos.
Margem do nervo óptico positiva ou infiltração extraescleral são indicações absolutas para terapia pós-operatória, realizando quimioterapia sistêmica e radioterapia. Infiltração coroidal acentuada ou infiltração do nervo óptico além da lâmina cribrosa são avaliadas como fatores de risco relativos para metástase.
Condições para preservação do globo ocular
T1 (lesão intraocular inicial): Taxa de preservação >90%
Diâmetro do tumor ≤3 mm: Laserterapia ou criocoagulação como primeira escolha
Espessura do tumor ≤5 mm: Braquiterapia é indicada
Quimioterapia sistêmica → terapia local: Em casos avançados, tentativa de preservação após redução
Condições que requerem enucleação
T3 (infiltração extraocular): Taxa de preservação cerca de 10%
Infiltração da câmara anterior ou íris: Risco de disseminação extraocular
Quando a recuperação da função visual não é esperada: Prioridade ao prognóstico de vida
QExiste possibilidade de preservar o olho?
A
Em lesões intraoculares iniciais (T1), a preservação do olho é possível em mais de 90% dos casos. O tumor é reduzido com quimioterapia sistêmica (VEC) e depois consolidado com laser, criocoagulação ou braquiterapia. Em casos avançados (T3), a taxa de preservação ocular é de cerca de 10%, podendo ser necessária a enucleação. A escolha do tratamento é determinada pelo especialista com base no estágio da doença e na expectativa de função visual.
6. Fisiopatologia e Mecanismo Detalhado de Ocorrência
O gene RB1 localizado no braço longo do cromossomo 13 (13q14.2) codifica a proteína RB1 (pRb), que desempenha um papel importante na regulação da divisão celular. A pRb se liga ao fator de transcrição E2F e inibe a transição G1/S do ciclo celular, controlando assim a proliferação celular como uma proteína supressora de tumor.
De acordo com a teoria dos dois eventos proposta por Knudson, a malignidade ocorre quando ambos os alelos do gene RB1 em uma única célula são inativados.
Hereditário: O primeiro evento (mutação germinativa) está presente em todas as células. Quando o segundo evento (mutação somática, LOH, etc.) ocorre em uma célula da retina, forma-se o tumor. Portanto, os tumores tendem a ser bilaterais e múltiplos, e são diagnosticados relativamente cedo.
Não hereditário: Tanto o primeiro quanto o segundo evento ocorrem como mutações somáticas na mesma célula da retina. Como a probabilidade de ambos os eventos ocorrerem por acaso em uma única célula é baixa, os tumores são frequentemente unilaterais e solitários, e o diagnóstico tende a ser mais tardio em comparação com os casos hereditários.
Em casos hereditários, o primeiro evento do RB1 está presente em todas as células do corpo. Quando o segundo evento ocorre em células não retinianas (como ossos, tecidos moles), desenvolve-se um tumor maligno primário secundário. O osteossarcoma é o mais comum, ocorrendo frequentemente após a adolescência. O risco de câncer secundário aumenta ainda mais em casos hereditários que receberam radioterapia externa, portanto, a radioterapia externa é atualmente de uso limitado.
A IAC (quimioterapia intra-arterial) administra melfalan diretamente na artéria oftálmica, proporcionando alta concentração do fármaco intraocular com toxicidade sistêmica mínima. Pode ampliar as chances de preservação ocular mesmo em casos avançados com disseminação vítrea. Atualmente, a avaliação de resultados a longo prazo em grandes coortes está em andamento.
A injeção intravítrea de melfalan é realizada para tratar disseminação vítrea, mas a padronização internacional de protocolos de dose e intervalo é um desafio. Várias séries de casos relatam altas taxas de controle da disseminação, e espera-se que ensaios prospectivos futuros estabeleçam seu papel.
O estudo de coorte holandês mostrou correlação clara entre a realização de rastreamento completo e o diagnóstico precoce (mediana de 18 dias), quantificando os efeitos adversos da interrupção do rastreamento 2). A otimização de protocolos baseada em estratificação de risco também está em andamento, como a proposta de redução da idade de término do rastreamento (2 anos) para grupos de baixo risco 2). A disseminação global das recomendações da AAOOP e a padronização de protocolos regionais são desafios futuros 1).
Embora raro, há relatos de casos de retinoblastoma concomitante com malformações cerebrais congênitas ou anomalias cromossômicas. Um relato de retinoblastoma bilateral com síndrome de Dandy-Walker destacou a importância da avaliação sistêmica, incluindo o background do desenvolvimento neurológico, além dos sintomas oculares. 4)
Com a disseminação do sequenciamento de próxima geração (NGS), a sensibilidade para detectar mutações germinativas do RB1 aumentou. A possibilidade de monitoramento usando biópsia líquida (DNA tumoral circulante no sangue) também está sendo estudada, com potencial aplicação na estratificação prognóstica sem biópsias invasivas.
Há necessidade de padronização de protocolos de vigilância de câncer secundário para sobreviventes de longo prazo de retinoblastoma hereditário. Especialmente quanto à frequência e momento de término do rastreamento multiórgãos usando RM de corpo inteiro, evidências estão sendo acumuladas por meio de estudos de coorte contínuos.
A injeção arterial oftálmica seletiva (IAC) e a quimioterapia intravítrea (IVitC) estão sendo cada vez mais reconhecidas internacionalmente como terapias que aumentam as taxas de preservação ocular em casos avançados ou com disseminação vítrea. Em revisões abrangentes de terapia conservadora, são posicionadas como estratégias para expandir a preservação ocular sem piorar o risco de metástase, e os resultados são influenciados pela adoção em cada país e pela centralização de instalações especializadas. 5)
Disparidade de Tratamento em Países em Desenvolvimento
Embora a taxa de sobrevida em 5 anos nos países desenvolvidos seja superior a 95%, relatos indicam que em países de baixa e média renda na África e Ásia ela fica entre 25% e 70%. Revisões sistemáticas e análises por nível de renda do país mostram consistentemente disparidades nas taxas de sobrevida e preservação ocular, sendo os principais fatores o diagnóstico tardio, acesso insuficiente aos cuidados de saúde e falta de instalações especializadas. A disseminação de programas de triagem comunitários, incluindo o método do reflexo vermelho, é um desafio internacional. 6, 7)
Skalet AH, Gombos DS, Gallie BL, Kim JW, Shields CL, Marr BP, Plon SE, Chévez-Barrios P. Screening Children at Risk for Retinoblastoma: Consensus Report from the American Association of Ophthalmic Oncologists and Pathologists. Ophthalmology. 2018;125(3):453-458. doi:10.1016/j.ophtha.2017.09.001. PMID:29056300.
Badalova NA, van Hoefen Wijsard M, Dommering CJ, et al. At what age could screening for familial retinoblastoma be stopped? Revised Dutch retrospective population-based cohort study 1991-2019. Ophthalmology. 2024;131(10):1189-1196.
Jakati S, Vempuluru VS, Kaliki S. Intraocular Cysticercosis Masquerading as Cavitary Retinoblastoma. Ophthalmology. 2025;132(4):e68. doi:10.1016/j.ophtha.2024.05.017. PMID:38878044.
Lomi N, Das D, Chawla B, Parampalli Ravindra A. Retinoblastoma in Dandy-Walker Syndrome. Cureus. 2025;17(8):e89663. doi:10.7759/cureus.89663. PMID:40926918; PMCID:PMC12415497.
Francis L. Munier, Maja Beck-Popovic, Guillermo L. Chantada, David Cobrinik, Tero T. Kivelä, Dietmar Lohmann, Philippe Maeder, Annette C. Moll, et al. Conservative management of retinoblastoma: Challenging orthodoxy without compromising the state of metastatic grace. “Alive, with good vision and no comorbidity”. Progress in Retinal and Eye Research. 2019;73:100764. doi:10.1016/j.preteyeres.2019.05.005.
Wong ES, Choy RW, Zhang Y, et al. Global retinoblastoma survival and globe preservation: a systematic review and meta-analysis. Lancet Glob Health. 2022;10(3):e380-e389.
Global Retinoblastoma Study Group, Fabian ID, Abdallah E, Abdullahi SU, Abdulqader RA, Adamou Boubacar S, Ademola-Popoola DS, Adio A, Afshar AR, Aggarwal P, Aghaji AE, Ahmad A, Akib MNR, Al Harby L, Al Ani MH, Alakbarova A, Portabella SA, Al-Badri SAF, Alcasabas APA, Al-Dahmash SA, Alejos A, Alemany-Rubio E, Alfa Bio AI, Alfonso Carreras Y, Al-Haddad C, Al-Hussaini HHY, Ali AM, Alia DB, Al-Jadiry MF, Al-Jumaily U, Alkatan HM, All-Eriksson C, Al-Mafrachi AARM, Almeida AA, Alsawidi KM, Al-Shaheen AASM, Al-Shammary EH, Amiruddin PO, Antonino R, Astbury NJ, Atalay HT, Atchaneeyasakul LO, Atsiaya R, Attaseth T, Aung TH, Ayala S, Baizakova B, Balaguer J, Balayeva R, Balwierz W, Barranco H, Bascaran C, Beck Popovic M, Benavides R, Benmiloud S, Bennani Guebessi N, Berete RC, Berry JL, Bhaduri A, Bhat S, Biddulph SJ, Biewald EM, Bobrova N, Boehme M, Boldt HC, Bonanomi MTBC, Bornfeld N, Bouda GC, Bouguila H, Boumedane A, Brennan RC, Brichard BG, Buaboonnam J, Calderón-Sotelo P, Calle Jara DA, Camuglia JE, Cano MR, Capra M, Cassoux N, Castela G, Castillo L, Català-Mora J, Chantada GL, Chaudhry S, Chaugule SS, Chauhan A, Chawla B, Chernodrinska VS, Chiwanga FS, Chuluunbat T, Cieslik K, Cockcroft RL, Comsa C, Correa ZM, Correa Llano MG, Corson TW, Cowan-Lyn KE, Csóka M, Cui X, Da Gama IV, Dangboon W, Das A, Das S, Davanzo JM, Davidson A, De Potter P, Delgado KQ, Demirci H, Desjardins L, Diaz Coronado RY, Dimaras H, Dodgshun AJ, Donaldson C, Donato Macedo CR, Dragomir MD, Du Y, Du Bruyn M, Edison KS, Eka Sutyawan IW, El Kettani A, Elbahi AM, Elder JE, Elgalaly D, Elhaddad AM, Elhassan MMA, Elzembely MM, Essuman VA, Evina TGA, Fadoo Z, Fandiño AC, Faranoush M, Fasina O, Fernández DDPG, Fernández-Teijeiro A, Foster A, Frenkel S, Fu LD, Fuentes-Alabi SL, Gallie BL, Gandiwa M, Garcia JL, García Aldana D, Gassant PY, Geel JA, Ghassemi F, Girón AV, Gizachew Z, Goenz MA, Gold AS, Goldberg-Lavid M, Gole GA, Gomel N, Gonzalez E, Gonzalez Perez G, González-Rodríguez L, Garcia Pacheco HN, Graells J, Green L, Gregersen PA, Grigorovski NDAK, Guedenon KM, Gunasekera DS, Gündüz AK, Gupta H, Gupta S, Hadjistilianou T, Hamel P, Hamid SA, Hamzah N, Hansen ED, Harbour JW, Hartnett ME, Hasanreisoglu M, Hassan S, Hassan S, Hederova S, Hernandez J, Hernandez LMC, Hessissen L, Hordofa DF, Huang LC, Hubbard GB, Hummlen M, Husakova K, Hussein Al-Janabi AN, Ida R, Ilic VR, Jairaj V, Jeeva I, Jenkinson H, Ji X, Jo DH, Johnson KP, Johnson WJ, Jones MM, Kabesha TBA, Kabore RL, Kaliki S, Kalinaki A, Kantar M, Kao LY, Kardava T, Kebudi R, Kepak T, Keren-Froim N, Khan ZJ, Khaqan HA, Khauv P, Kheir WJ, Khetan V, Khodabande A, Khotenashvili Z, Kim JW, Kim JH, Kiratli H, Kivelä TT, Klett A, Komba Palet JEK, Krivaitiene D, Kruger M, Kulvichit K, Kuntorini MW, Kyara A, Lachmann ES, Lam CPS, Lam GC, Larson SA, Latinovic S, Laurenti KD, Le BHA, Lecuona K, Leverant AA, Li C, Limbu B, Long QB, López JP, Lukamba RM, Lumbroso L, Luna-Fineman S, Lutfi D, Lysytsia L, Magrath GN, Mahajan A, Majeed AR, Maka E, Makan M, Makimbetov EK, Manda C, Martín Begue N, Mason L, Mason JO, Matende IO, Materin M, Mattosinho CCDS, Matua M, Mayet I, Mbumba FB, McKenzie JD, Medina-Sanson A, Mehrvar A, Mengesha AA, Menon V, Mercado GJVD, Mets MB, Midena E, Mishra DKC, Mndeme FG, Mohamedani AA, Mohammad MT, Moll AC, Montero MM, Morales RA, Moreira C, Mruthyunjaya P, Msina MS, Msukwa G, Mudaliar SS, Muma KI, Munier FL, Murgoi G, Murray TG, Musa KO, Mushtaq A, Mustak H, Muyen OM, Naidu G, Nair AG, Naumenko L, Ndoye Roth PA, Nency YM, Neroev V, Ngo H, Nieves RM, Nikitovic M, Nkanga ED, Nkumbe H, Nuruddin M, Nyaywa M, Obono-Obiang G, Oguego NC, Olechowski A, Oliver SCN, Osei-Bonsu P, Ossandon D, Paez-Escamilla MA, Pagarra H, Painter SL, Paintsil V, Paiva L, Pal BP, Palanivelu MS, Papyan R, Parrozzani R, Parulekar M, Pascual Morales CR, Paton KE, Pawinska-Wasikowska K, Pe’er J, Peña A, Peric S, Pham CTM, Philbert R, Plager DA, Pochop P, Polania RA, Polyakov VG, Pompe MT, Pons JJ, Prat D, Prom V, Purwanto I, Qadir AO, Qayyum S, Qian J, Rahman A, Rahman S, Rahmat J, Rajkarnikar P, Ramanjulu R, Ramasubramanian A, Ramirez-Ortiz MA, Raobela L, Rashid R, Reddy MA, Reich E, Renner LA, Reynders D, Ribadu D, Riheia MM, Ritter-Sovinz P, Rojanaporn D, Romero L, Roy SR, Saab RH, Saakyan S, Sabhan AH, Sagoo MS, Said AMA, Saiju R, Salas B, San Román Pacheco S, Sánchez GL, Sayalith P, Scanlan TA, Schefler AC, Schoeman J, Sedaghat A, Seregard S, Seth R, Shah AS, Shakoor SA, Sharma MK, Sherief ST, Shetye NG, Shields CL, Siddiqui SN, Sidi Cheikh S, Silva S, Singh AD, Singh N, Singh U, Singha P, Sitorus RS, Skalet AH, Soebagjo HD, Sorochynska T, Ssali G, Stacey AW, Staffieri SE, Stahl ED, Stathopoulos C, Stirn Kranjc B, Stones DK, Strahlendorf C, Suarez MEC, Sultana S, Sun X, Sundy M, Superstein R, Supriyadi E, Surukrattanaskul S, Suzuki S, Svojgr K, Sylla F, Tamamyan G, Tan D, Tandili A, Tarrillo Leiva FF, Tashvighi M, Tateshi B, Tehuteru ES, Teixeira LF, Teh KH, Theophile T, Toledano H, Trang DL, Traoré F, Trichaiyaporn S, Tuncer S, Tyau-Tyau H, Umar AB, Unal E, Uner OE, Urbak SF, Ushakova TL, Usmanov RH, Valeina S, van Hoefen Wijsard M, Varadisai A, Vasquez L, Vaughan LO, Veleva-Krasteva NV, Verma N, Victor AA, Viksnins M, Villacís Chafla EG, Vishnevskia-Dai V, Vora T, Wachtel AE, Wackernagel W, Waddell K, Wade PD, Wali AH, Wang YZ, Weiss A, Wilson MW, Wime ADC, Wiwatwongwana A, Wiwatwongwana D, Wolley Dod C, Wongwai P, Xiang D, Xiao Y, Yam JC, Yang H, Yanga JM, Yaqub MA, Yarovaya VA, Yarovoy AA, Ye H, Yousef YA, Yuliawati P, Zapata López AM, Zein E, Zhang C, Zhang Y, Zhao J, Zheng X, Zhilyaeva K, Zia N, Ziko OAO, Zondervan M, Bowman R.. Global Retinoblastoma Presentation and Analysis by National Income Level. JAMA Oncol. 2020;6(5):685-695. doi:10.1001/jamaoncol.2019.6716. PMID:32105305; PMCID:PMC7047856.
Copie o texto do artigo e cole no assistente de IA de sua preferência.
Artigo copiado para a área de transferência
Abra um assistente de IA abaixo e cole o texto copiado na conversa.