رتینوبلاستوما یک تومور بدخیم شبکیه در نوزادان و کودکان خردسال است. این بیماری ناشی از تبدیل سلولهای نابالغ شبکیه به سلولهای سرطانی و تشکیل توده است و به عنوان یک بیماری تکژنی ناشی از جهش در ژن RB1 واقع در بازوی بلند کروموزوم 13 (13q14.2) در نظر گرفته میشود. تفاوت جنسیتی ندارد و ۹۵٪ موارد تا سن ۵ سالگی تشخیص داده میشود.
فراوانی بروز ۱ در ۱۵٬۰۰۰ تا ۲۳٬۰۰۰ تولد است و در ژاپن سالانه ۷۰ تا ۸۰ نفر مبتلا میشوند. نسبت موارد یکطرفه به دوطرفه ۳:۲ است و زمان تشخیص در موارد یکطرفه به طور متوسط ۲۱ ماه و در موارد دوطرفه ۸ ماه است که نشاندهنده تشخیص زودهنگام در موارد دوطرفه است. بقای ۵ ساله در کشورهای توسعهیافته برای موارد محدود به داخل چشم بیش از ۹۵٪ و مطلوب است.
رتینوبلاستوما بر اساس نوع جهش ژنتیکی به دو دسته اصلی تقسیم میشود.
طبقهبندی
نوع جهش
ویژگیهای بالینی
خطر ارثی
ارثی (جهش زایا)
جهش RB1 در سلولهای زایا
معمولاً دوطرفه و چندتوموری
۵۰٪ احتمال انتقال به فرزندان
غیرارثی (جهش جسمی)
جهش جسمی در یک سلول شبکیه
یکطرفه و تکتوموری
بدون خطر ارث برای نسل بعد
ارثی (جهش زایا): در این حالت، اولین جهش در تمام سلولهای بدن وجود دارد. با وقوع جهش دوم، سرطان ایجاد میشود. احتمال بروز تومورهای دوطرفه و چندگانه زیاد است و ۵۰٪ از فرزندان به ارث میبرند. خطر ابتلا به سرطانهای ثانویه مانند استئوسارکوم (۱۵٫۷٪ در ۲۰ سال) وجود دارد.
غیرارثی (جهش جسمی): زمانی رخ میدهد که در یک سلول شبکیه، هر دو الل ژن RB1 دچار جهش شوند. معمولاً به صورت یکطرفه و تکتوموری ظاهر میشود و خطری برای نسل بعد ندارد.
با این حال، حتی در موارد تکچشمی نیز برخی شامل جهشهای RB1 در سلولهای زایا هستند. بنابراین، صرفاً به دلیل تکچشمی بودن، وراثت را رد نکنید و سابقه خانوادگی، سن شروع و تعداد تومورها را با در نظر گرفتن مشاوره ژنتیک و ارزیابی ژنتیکی تفسیر کنید. 1)
مرحلهبندی بیماری مستقیماً با استراتژی درمان حفظ چشم مرتبط است.
مرحله
وضعیت ضایعه
برآورد میزان حفظ چشم
T1 (ضایعه اولیه داخل چشمی)
محدود به داخل چشم، بدون پیشرفت
بیش از 90٪
T2 (ضایعه پیشرفته داخل چشمی)
پیشرفت داخل چشمی
حدود 50٪
T3
ضایعه پیشرفته با تهاجم خارج چشمی
حدود 10٪
Qآیا رتینوبلاستوما ارثی است؟
A
حدود 40% موارد ارثی (جهش RB1 در سلولهای زایا) هستند و با احتمال 50% به فرزندان منتقل میشوند. حدود 60% باقیمانده غیرارثی (فقط جهش سوماتیک) بوده و خطر ارثی برای نسل بعد ندارند. موارد ارثی تمایل به دوطرفه و چندکانونی دارند. انجام آزمایش ژنتیک در زمان تشخیص و دریافت مشاوره ژنتیک توصیه میشود.
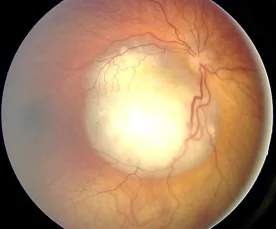
Aerts I, et al. Retinoblastoma. Orphanet J Rare Dis. 2006 Aug 25; 1:31. Figure 2. PMCID: PMC1586012. License: CC BY.
تصویر فوندوس رتینوبلاستوما به صورت ضایعه برجسته سفید با عروق فراوان مشاهده میشود که نمایانگر یافته معمول ایجادکننده لوکوکوریا (سفیدی مردمک) است. این تصویر مربوط به یافتههای فوندوسکوپی و لوکوکوریا است که در بخش «2. علائم اصلی و یافتههای بالینی» بحث شده است.
در بسیاری از موارد، تومور داخل چشم بزرگ شده و به صورت لوکوکوریا (سفیدی مردمک) تشخیص داده میشود. اگر تومور در ناحیه ماکولا ایجاد شود، به دلیل دید ضعیف باعث استرابیسم (انحراف چشم) شده و ممکن است از این طریق تشخیص داده شود. در کودکان بزرگتر، کاهش بینایی خودآگاه و در کودکان خردسال، مالش چشم مبتلا به دید ضعیف میتواند علامت اولیه باشد.
علائم اولیه
لوکوکوریا (سفیدی مردمک): شایعترین علامت اولیه. تومور داخل چشم بزرگ شده و مردمک سفید به نظر میرسد.
استرابیسم (انحراف چشم): ناشی از دید ضعیف به دلیل تومور در ماکولا. چشم با دید ضعیف به سمت خارج منحرف میشود.
کاهش بینایی خودآگاه: در کودکان بزرگتر مشاهده میشود.
مالش چشم: در کودکان خردسال با چشم دارای دید ضعیف دیده میشود.
علائم مرحله پیشرفته
کدورت قرنیه و افزایش فشار داخل چشم: ناشی از فشار تومور به عدسی یا گلوکوم نئوواسکولار (NVG) که منجر به افزایش فشار داخل چشم میشود.
قرمزی ملتحمه و تورم پلک: به عنوان یافته التهابی مشاهده میشود.
درد: به دلیل افزایش فشار داخل چشم یا نکروز تومور ایجاد میشود.
برجستگی چشم (اگزوفتالمی): هنگامی که تومور به خارج از چشم گسترش مییابد، مشاهده میشود.
ضایعات برجسته سفید رنگ عروقی مشاهده میشود و در صورت همراهی با کلسیفیکاسیون، تشخیص قطعی آسانتر است. اغلب با انتشار سلولهای توموری در زجاجیه (ویتره) همراه است.
روش رفلکس قرمز اساس غربالگری بیماریهای چشمی در نوزادان و شیرخواران است. اگر اندازه مردمکهای هر دو چشم برابر و بازتاب آن زرد-نارنجی روشن و متقارن باشد، طبیعی است. اگر رفلکس تیره یا بیش از حد روشن باشد، یا بین دو چشم تفاوت وجود داشته باشد، غیرطبیعی تلقی شده و نیاز به بررسی بیشتر دارد.
Qآیا مردمک سفید همیشه به معنای رتینوبلاستوما است؟
A
علل مردمک سفید علاوه بر رتینوبلاستوما شامل موارد متعددی مانند باقیمانده عروق جنینی (هیپرپلازی زجاجیه اولیه)، رتینوپاتی نوزادان نارس، بیماری کوتس و غیره است. با این حال، در صورت مشاهده مردمک سفید، مراجعه فوری به چشمپزشک و رد رتینوبلاستوما در اولویت اول قرار دارد. تأخیر در تشخیص مستقیماً بر پیشآگهی تأثیر میگذارد، بنابراین در صورت شک، ارجاع در همان روز توصیه میشود.
جهش در ژن RB1 واقع در بازوی بلند کروموزوم 13 (13q14.2) علت این بیماری است. ژن RB1 پروتئین RB1 (پروتئین رتینوبلاستوما) را تولید میکند که نقش مهمی در کنترل تقسیم سلولی دارد.
در هر سلول دو نسخه از ژن وجود دارد. جهش در یک نسخه به تنهایی عملکرد سلول را حفظ میکند، اما جهش در هر دو نسخه باعث از دست رفتن کنترل تقسیم سلولی و بدخیمی میشود (نظریه دو مرحلهای سرطانزایی، فرضیه نادسون).
نوع ارثی (جهش زاینده): اولین جهش در سلولهای زاینده (در تمام سلولهای بدن وجود دارد). هنگامی که جهش دوم در یک سلول جسمی رخ دهد، سرطان ایجاد میشود. احتمال بروز تومورهای دوطرفه و چندگانه زیاد است.
نوع غیرارثی (جهش جسمی): هر دو جهش اول و دوم در یک سلول جسمی رخ میدهد. به صورت تومور یک طرفه و منفرد ظاهر میشود.
در موارد ارثی باید به خطر سرطان ثانویه توجه کرد. استئوسارکوم نمونهای است و در موارد ارثی در 15.7% موارد طی 20 سال رخ میدهد. سرطان ثانویه اغلب پس از دهه دوم زندگی بروز میکند.
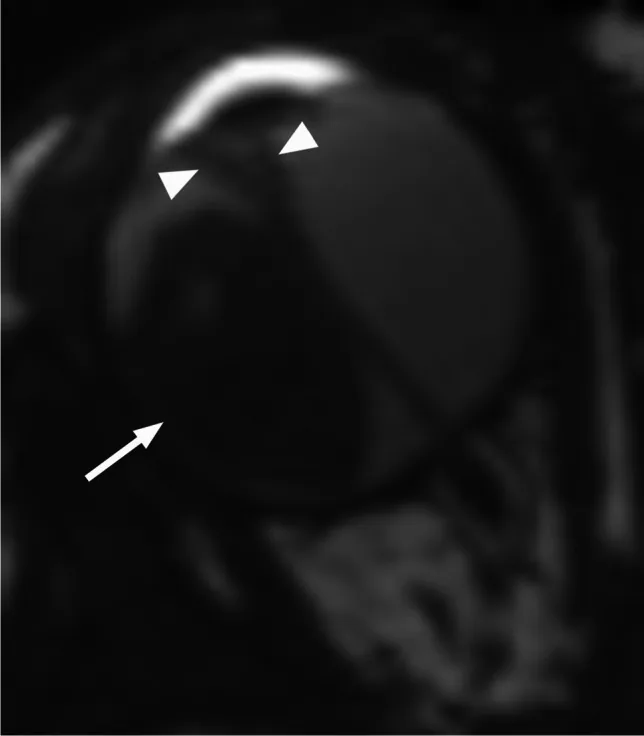
Rumboldt Z, Dodig D, Galluzzi P, et al. Retinoblastoma and beyond: pediatric orbital mass lesions. Neuroradiology. 2025;67(2):469-492. Figure 2. PMID: 39729290; PMCID: PMC11893699; DOI: 10.1007/s00234-024-03517-6. License: CC BY.
تصویر MRI محوری با وزن T2 که رتینوبلاستومای اگزوفیتیک (فلش) را با جداشدگی ثانویه V شکل شبکیه (سر فلش) نشان میدهد. این تصویر مربوط به الگوهای رشد اگزوفیتیک و اندوفیتیک و یافتههای MRI است که در بخش «4. تشخیص و روشهای آزمایش» بحث شده است.
بیوپسی تومور داخل چشمی انجام نمیشود. ضایعات داخل چشمی از طریق بافت شفاف مستقیماً قابل مشاهده هستند و دقت تشخیص بالینی بالاست. همچنین، بیوپسی تومور داخل چشمی میتواند باعث انتشار سلولهای توموری به خارج از کره چشم و خطر متاستاز شود. در صورت درمان حفظ کره چشم، درمان بر اساس تشخیص بالینی آغاز میشود.
تشخیص افتراقی بیماریهایی که باعث لکوسوری (رفلکس سفید مردمک) میشوند، بسیار مهم است.
باقیمانده عروق جنینی (باقیمانده زجاجیه اولیه): بررسی وجود تومور جامد با سونوگرافی
رتینوپاتی نوزادان نارس: سابقه زایمان زودرس و وزن کم هنگام تولد سرنخی برای تشخیص افتراقی است
بیماری کوتس: تجمع ترشحات زرد-سفید زیر شبکیه. الگوی عروق تومور متفاوت است
همارتوم سلول ستارهای: وجود عروق تومور، محل ضایعه در OCT و روند رشد برای تشخیص افتراقی استفاده میشود
سیستیسرکوز چشمی: اگرچه نادر است، میتواند Rb را تقلید کند. موردی از یک پسر ۴ ساله با مردمک سفید که به ظن Rb انوکلئاسیون شد و تشخیص پاتولوژی سیستیسرکوز بود گزارش شده است3)
برای غربالگری کودکان با سابقه خانوادگی Rb، توصیه AAOOP 2018 به طور گسترده در سطح بینالمللی مورد استفاده قرار میگیرد1).
خطر
برنامه غربالگری
زمان پایان
بالا (>7.5%)
تولد تا ۸ هفته: هر ۲-۴ هفته → ۸-۱۲ هفته: ماهانه → ۱-۲ سال: هر ۲ ماه → ۲-۳ سال: هر ۳ ماه → ۳-۴ سال: هر ۴ ماه → ۴-۷ سال: هر ۶ ماه
۷ سال (ناقلان جهش RB1 مادامالعمر)
متوسط (۱-۷.۵%)
تولد تا ۳ ماه: ماهانه → کاهش تدریجی
۷ سال
کم (<1%)
تولد تا 3 ماه: ماهی یک بار → کاهش تدریجی
7 سال
یک مطالعه کوهورت گذشتهنگر از دادههای ملی هلند (1991-2019، 38 نفر از 332 نفر مبتلا به Rb خانوادگی) نشان داد که 28 نفری که غربالگری کامل دریافت کردند، همگی قبل از 1 سالگی (میانه 18 روز) تشخیص داده شدند، در حالی که در 10 نفر با غربالگری ناقص، تشخیص به طور قابل توجهی با تاخیر (میانه 420 روز، محدوده 59 روز تا 4.8 سال) انجام شد2). همچنین اصلاح پروتکلی برای کاهش سن پایان غربالگری در گروه کمخطر (<3%) به 2 سال پیشنهاد شده است2).
در موارد خانوادگی، ادامه غربالگری فوندوس از بدو تولد به طور مستقیم با پیشآگهی مرتبط است. حتی در مطالعات ثبت کلاسیک، زمان تشخیص موارد خانوادگی ارتباط نزدیکی با دفعات غربالگری دارد و در حال حاضر، با ترکیب وجود یا عدم وجود جهش RB1، سن پایان غربالگری به صورت فردی تعیین میشود.1, 2)
Qاگر در خانواده سابقه رتینوبلاستوما وجود داشته باشد، غربالگری کودک تا چه زمانی ضروری است؟
A
توصیه AAOOP معاینه منظم فوندوس تا 7 سالگی است. در صورت انجام غربالگری کامل، اکثر موارد قبل از 1 سالگی تشخیص داده میشوند. اگر آزمایش ژنتیکی خطر جهش RB1 را رد کند، میتوان غربالگری را زودتر پایان داد. برای ناقلان جهش RB1، پیگیری نامنظم هر 1-2 سال پس از 7 سالگی توصیه میشود.
در مراحل اولیه داخل چشمی که انتظار حفظ عملکرد بینایی وجود دارد، درمان حفظ کننده چشم به طور فعال انجام میشود. در مراحل پیشرفته داخل چشمی، اغلب انتظار حفظ عملکرد بینایی وجود ندارد، اما در صورت تمایل خانواده، درمان حفظ کننده چشم در نظر گرفته میشود. درمان نیاز به تخصص بالایی دارد و ارجاع زودهنگام به مراکز تخصصی مهم است.
هدف تومورهایی با قطر تا حدود 3 میلیمتر است. تابش مستقیم لیزر مادون قرمز میتواند کنترل موضعی حدود 90٪ را فراهم کند. در صورت وجود تومور در ناحیه ماکولا، برای جلوگیری از اختلال غیرقابل برگشت عملکرد بینایی، توصیه میشود ابتدا شیمیدرمانی سیستمیک انجام شود.
② کرایوتراپی (انجماد)
هدف تومورهایی با قطر حدود 3 میلیمتر در ناحیه استوا یا محیطتر است. روش رایج، روش انجماد-ذوب سهگانه (triple freeze-thaw) است که انجماد و ذوب را سه بار تکرار میکند و مانند لیزر، کنترل موضعی حدود 90٪ را فراهم میکند.
③ براکیتراپی (پرتو درمانی موضعی)
این روش برای تومورهای موضعی با ضخامت ≤5 میلیمتر و قطر ≤15 میلیمتر که از دیسک بینایی فاصله دارند، مناسب است. در ژاپن و اروپا از ¹⁰⁶Ru (روتنیم-106، منبع بتا) و در آمریکای شمالی از منبع ¹²⁵I استفاده میشود. در این درمان، یک منبع پرتوزا به طور موقت بر روی صلبیه در محل تومور دوخته میشود. این روش نیاز به اتاق درمان ویژه دارد و در مراکز محدودی انجام میشود. میزان کنترل موضعی 80 تا 90 درصد است.
④ شیمیدرمانی سیستمیک (رژیم VEC)
این درمان به عنوان خط اول برای تومورهای داخل چشمی پیشرفته استفاده میشود. شیمیدرمانی ترکیبی با سه دارو به طور گسترده انجام میشود، اما میزان بهبودی با درمان به تنهایی کمتر از 10 درصد است. پس از کوچک شدن تومور، درمان موضعی (لیزر، کرایوتراپی، براکیتراپی) برای تثبیت انجام میشود.
دارو
دوز (بر اساس سطح بدن)
دوز (بر اساس وزن برای کودکان ≤36 ماه)
برنامه تجویز
وینکریستین (Oncovin®)
1.5 mg/m²
0.05 mg/kg
روز 1
کربوپلاتین (Paraplatin®)
560 mg/m²
18.6 mg/kg
روز 1
اتوپوزید (Vepesid®)
150 mg/m²
5 mg/kg
روز 1 و 2
هر 3 تا 4 هفته، 2 تا 6 بار تکرار شود (همگی به صورت تزریق وریدی).
با استفاده از کاتتر، دارو (ملفالان؛ محلول تزریقی Alkeran®) مستقیماً به شریان چشمی تزریق میشود. با رساندن مقدار بیشتری دارو به ناحیه موضعی چشم و کاهش دوز سیستمیک، عوارض جانبی مانند سرکوب مغز استخوان کاهش مییابد. این درمان تحت پوشش بیمه نیست، اما در بیش از 20 کشور جهان به عنوان یک درمان تحقیقاتی انجام میشود.
برای انتشار زجاجیهای، از آنجایی که اثر شیمیدرمانی سیستمیک و تزریق شریانی محدود است، تزریق داخل زجاجیهای ملفالان (محلول تزریقی Alkeran®) به صورت ترکیبی استفاده میشود. این یک درمان تحقیقاتی و خارج از پوشش بیمه است.
⑦ پرتودرمانی خارجی
پرتو ایکس با دوز 40 تا 46 گری به صورت کسری تابانده میشود. تا دهه 1990، این روش اصلی درمان حفظ چشم بود، اما با مشخص شدن تغییر شکل استخوان حدقه و افزایش سرطانهای ثانویه، اکنون تنها در مواردی که با سایر درمانها قابل کنترل نباشد، انجام میشود.
حاشیه مثبت عصب بینایی و تهاجم خارج صلبیه نشانههای مطلق برای درمانهای پس از جراحی هستند و شیمیدرمانی سیستمیک و پرتودرمانی انجام میشود. تهاجم قابل توجه به مشیمیه یا تهاجم عصب بینایی فراتر از صفحه کریبریفرم به عنوان عوامل خطر نسبی برای متاستاز ارزیابی میشوند.
شرایط حفظ چشم
T1 (ضایعه اولیه داخل چشمی): میزان حفظ بیش از 90٪
قطر تومور 3 میلیمتر یا کمتر: لیزر درمانی یا کرایوتراپی گزینه اول
ضخامت تومور 5 میلیمتر یا کمتر: براکیتراپی مناسب است
شیمیدرمانی سیستمیک → درمان موضعی: حتی در موارد پیشرفته، پس از کوچک شدن تومور، حفظ چشم تلاش میشود
شرایط نیاز به تخلیه چشم
T3 (تهاجم خارج چشمی): میزان حفظ حدود 10٪
گلوکوم نئوواسکولار (NVG): در صورت عدم کنترل فشار چشم
تهاجم به اتاق قدامی یا عنبیه: خطر انتشار خارج چشمی
زمانی که بهبود عملکرد بینایی انتظار نمیرود: اولویت با پیشآگهی حیاتی
Qآیا امکان حفظ چشم وجود دارد؟
A
در ضایعات اولیه داخل چشمی (T1)، حفظ چشم در بیش از 90% موارد امکانپذیر است. با شیمیدرمانی سیستمیک (رژیم VEC) تومور کوچک شده و با لیزر، کرایوتراپی و براکیتراپی تثبیت میشود. در موارد پیشرفته (T3)، میزان حفظ چشم حدود 10% است و ممکن است نیاز به تخلیه چشم باشد. انتخاب روش درمان بر اساس مرحله بیماری و چشمانداز عملکرد بینایی توسط پزشک متخصص تعیین میشود.
ژن RB1 واقع در بازوی بلند کروموزوم 13 (13q14.2) پروتئین RB1 (pRb) را کد میکند که نقش مهمی در کنترل تقسیم سلولی دارد. pRb با اتصال به فاکتور رونویسی E2F از انتقال G1/S در چرخه سلولی جلوگیری کرده و به عنوان یک پروتئین سرکوبگر تومور، رشد سلولی را کنترل میکند.
بر اساس فرضیه دو مرحلهای سرطانزایی که توسط نادسون مطرح شد، غیرفعال شدن هر دو الل ژن RB1 در یک سلول منجر به بدخیمی میشود.
ارثی: اولین جهش (جهش خط زایا) در تمام سلولها وجود دارد. با وقوع جهش دوم (جهش سوماتیک، LOH و غیره) در یکی از سلولهای شبکیه، سرطان ایجاد میشود. بنابراین تومورهای دوطرفه و چندگانه شایعتر بوده و نسبتاً زود تشخیص داده میشوند.
غیرارثی: هر دو جهش اول و دوم به صورت جهشهای سوماتیک در یک سلول شبکیه رخ میدهند. احتمال تصادفی بودن هر دو جهش در یک سلول کم است، بنابراین تومورها اغلب یک طرفه و منفرد بوده و تشخیص دیرتر از نوع ارثی صورت میگیرد.
در موارد ارثی، اولین جهش RB1 در تمام سلولهای بدن وجود دارد. اگر جهش دوم در سلولهای غیر از شبکیه (مانند استخوان، بافت نرم و غیره) رخ دهد، تومور بدخیم اولیه ثانویه ایجاد میشود. استئوسارکوم شایعترین است و اغلب پس از دهه دوم زندگی بروز میکند. در موارد ارثی که پرتودرمانی خارجی دریافت کردهاند، خطر سرطان ثانویه بیشتر افزایش مییابد، بنابراین امروزه پرتودرمانی خارجی به صورت محدود استفاده میشود.
IAC (شیمیدرمانی داخل شریانی) با تزریق مستقیم ملفالان به شریان چشمی، تحویل غلظت بالای دارو به داخل کره چشم را با حداقل سمیت سیستمیک ممکن میسازد. حتی در موارد پیشرفته با انتشار زجاجیهای، فرصت حفظ چشم را افزایش میدهد. در حال حاضر، ارزیابی نتایج بلندمدت در گروههای بزرگ در حال انجام است.
برای مقابله با انتشار زجاجیهای، تزریق داخل زجاجیهای ملفالان انجام میشود، اما استانداردسازی پروتکل بینالمللی دوز و فواصل تجویز یک چالش است. در چندین سری موارد، نرخ بالای کنترل انتشار گزارش شده است و انتظار میرود که کارآزماییهای آیندهنگر آینده جایگاه آن را تثبیت کنند.
در مطالعه کوهورت هلند، همبستگی واضحی بین شرکت در غربالگری کامل و تشخیص زودهنگام (میانه ۱۸ روز) نشان داده شد و مضرات قطع غربالگری کمّی شد 2). همچنین، پیشنهادهایی برای کاهش سن پایان غربالگری در گروههای کمخطر (۲ سال) و بهینهسازی پروتکل بر اساس طبقهبندی خطر در حال پیشرفت است 2). انتشار جهانی توصیههای AAOOP و استانداردسازی پروتکلهای منطقهای چالشهای آینده هستند 1).
اگرچه نادر است، گزارشهایی از موارد رتینوبلاستوما همراه با ناهنجاریهای مادرزادی مغز یا ناهنجاریهای کروموزومی وجود دارد. در گزارش رتینوبلاستومای دوطرفه همراه با سندرم دندی واکر، اهمیت ارزیابی کامل سیستمیک شامل زمینه عصبی-رشدی علاوه بر علائم چشمی نشان داده شده است. 4)
با گسترش توالییابی نسل بعدی (NGS)، حساسیت تشخیص جهشهای RB1 خط زایا افزایش یافته است. امکان پایش با استفاده از بیوپسی مایع (DNA تومور در گردش خون) نیز در حال بررسی است و انتظار میرود که برای طبقهبندی پیشآگهی بدون نیاز به بیوپسی تهاجمی به کار رود.
استانداردسازی پروتکل مراقبت از سرطان ثانویه برای بازماندگان بلندمدت رتینوبلاستومای ارثی ضروری است. به ویژه در مورد فراوانی و زمان پایان غربالگری چندعضوی با استفاده از MRI کل بدن، تحقیقات کوهورت مداوم در حال جمعآوری شواهد است.
تزریق شریانی انتخابی چشم (IAC) و شیمیدرمانی داخل زجاجیهای (IVitC) به عنوان درمانهایی که نرخ حفظ چشم را در موارد پیشرفته و موارد انتشار زجاجیهای افزایش دادهاند، در سطح بینالمللی در حال تثبیت هستند. در مرورهای درمان محافظهکارانه، این روشها به عنوان استراتژیهایی برای گسترش حفظ چشم بدون افزایش خطر متاستاز معرفی شدهاند و وضعیت اجرا در کشورهای مختلف و تمرکز در مراکز تخصصی بر نتایج تأثیر میگذارد. 5)
گزارشهایی وجود دارد که نشان میدهد در حالی که نرخ بقای ۵ ساله در کشورهای توسعهیافته بیش از ۹۵٪ است، در کشورهای کمدرآمد و متوسط آفریقا و آسیا این نرخ تنها بین ۲۵ تا ۷۰٪ است. مرور سیستماتیک و تحلیل بر اساس سطح درآمد کشورها به طور مداوم نابرابری در بقا و نرخ حفظ چشم را نشان میدهد که عوامل اصلی آن تشخیص دیرهنگام، دسترسی ناکافی به مراقبتهای پزشکی و کمبود امکانات تخصصی ذکر شده است. گسترش برنامههای غربالگری مبتنی بر جامعه، از جمله روش رفلکس قرمز، یک چالش بینالمللی است. 6, 7)
Skalet AH, Gombos DS, Gallie BL, Kim JW, Shields CL, Marr BP, Plon SE, Chévez-Barrios P. Screening Children at Risk for Retinoblastoma: Consensus Report from the American Association of Ophthalmic Oncologists and Pathologists. Ophthalmology. 2018;125(3):453-458. doi:10.1016/j.ophtha.2017.09.001. PMID:29056300.
Badalova NA, van Hoefen Wijsard M, Dommering CJ, et al. At what age could screening for familial retinoblastoma be stopped? Revised Dutch retrospective population-based cohort study 1991-2019. Ophthalmology. 2024;131(10):1189-1196.
Jakati S, Vempuluru VS, Kaliki S. Intraocular Cysticercosis Masquerading as Cavitary Retinoblastoma. Ophthalmology. 2025;132(4):e68. doi:10.1016/j.ophtha.2024.05.017. PMID:38878044.
Lomi N, Das D, Chawla B, Parampalli Ravindra A. Retinoblastoma in Dandy-Walker Syndrome. Cureus. 2025;17(8):e89663. doi:10.7759/cureus.89663. PMID:40926918; PMCID:PMC12415497.
Francis L. Munier, Maja Beck-Popovic, Guillermo L. Chantada, David Cobrinik, Tero T. Kivelä, Dietmar Lohmann, Philippe Maeder, Annette C. Moll, et al. Conservative management of retinoblastoma: Challenging orthodoxy without compromising the state of metastatic grace. “Alive, with good vision and no comorbidity”. Progress in Retinal and Eye Research. 2019;73:100764. doi:10.1016/j.preteyeres.2019.05.005.
Wong ES, Choy RW, Zhang Y, et al. Global retinoblastoma survival and globe preservation: a systematic review and meta-analysis. Lancet Glob Health. 2022;10(3):e380-e389.
Global Retinoblastoma Study Group, Fabian ID, Abdallah E, Abdullahi SU, Abdulqader RA, Adamou Boubacar S, Ademola-Popoola DS, Adio A, Afshar AR, Aggarwal P, Aghaji AE, Ahmad A, Akib MNR, Al Harby L, Al Ani MH, Alakbarova A, Portabella SA, Al-Badri SAF, Alcasabas APA, Al-Dahmash SA, Alejos A, Alemany-Rubio E, Alfa Bio AI, Alfonso Carreras Y, Al-Haddad C, Al-Hussaini HHY, Ali AM, Alia DB, Al-Jadiry MF, Al-Jumaily U, Alkatan HM, All-Eriksson C, Al-Mafrachi AARM, Almeida AA, Alsawidi KM, Al-Shaheen AASM, Al-Shammary EH, Amiruddin PO, Antonino R, Astbury NJ, Atalay HT, Atchaneeyasakul LO, Atsiaya R, Attaseth T, Aung TH, Ayala S, Baizakova B, Balaguer J, Balayeva R, Balwierz W, Barranco H, Bascaran C, Beck Popovic M, Benavides R, Benmiloud S, Bennani Guebessi N, Berete RC, Berry JL, Bhaduri A, Bhat S, Biddulph SJ, Biewald EM, Bobrova N, Boehme M, Boldt HC, Bonanomi MTBC, Bornfeld N, Bouda GC, Bouguila H, Boumedane A, Brennan RC, Brichard BG, Buaboonnam J, Calderón-Sotelo P, Calle Jara DA, Camuglia JE, Cano MR, Capra M, Cassoux N, Castela G, Castillo L, Català-Mora J, Chantada GL, Chaudhry S, Chaugule SS, Chauhan A, Chawla B, Chernodrinska VS, Chiwanga FS, Chuluunbat T, Cieslik K, Cockcroft RL, Comsa C, Correa ZM, Correa Llano MG, Corson TW, Cowan-Lyn KE, Csóka M, Cui X, Da Gama IV, Dangboon W, Das A, Das S, Davanzo JM, Davidson A, De Potter P, Delgado KQ, Demirci H, Desjardins L, Diaz Coronado RY, Dimaras H, Dodgshun AJ, Donaldson C, Donato Macedo CR, Dragomir MD, Du Y, Du Bruyn M, Edison KS, Eka Sutyawan IW, El Kettani A, Elbahi AM, Elder JE, Elgalaly D, Elhaddad AM, Elhassan MMA, Elzembely MM, Essuman VA, Evina TGA, Fadoo Z, Fandiño AC, Faranoush M, Fasina O, Fernández DDPG, Fernández-Teijeiro A, Foster A, Frenkel S, Fu LD, Fuentes-Alabi SL, Gallie BL, Gandiwa M, Garcia JL, García Aldana D, Gassant PY, Geel JA, Ghassemi F, Girón AV, Gizachew Z, Goenz MA, Gold AS, Goldberg-Lavid M, Gole GA, Gomel N, Gonzalez E, Gonzalez Perez G, González-Rodríguez L, Garcia Pacheco HN, Graells J, Green L, Gregersen PA, Grigorovski NDAK, Guedenon KM, Gunasekera DS, Gündüz AK, Gupta H, Gupta S, Hadjistilianou T, Hamel P, Hamid SA, Hamzah N, Hansen ED, Harbour JW, Hartnett ME, Hasanreisoglu M, Hassan S, Hassan S, Hederova S, Hernandez J, Hernandez LMC, Hessissen L, Hordofa DF, Huang LC, Hubbard GB, Hummlen M, Husakova K, Hussein Al-Janabi AN, Ida R, Ilic VR, Jairaj V, Jeeva I, Jenkinson H, Ji X, Jo DH, Johnson KP, Johnson WJ, Jones MM, Kabesha TBA, Kabore RL, Kaliki S, Kalinaki A, Kantar M, Kao LY, Kardava T, Kebudi R, Kepak T, Keren-Froim N, Khan ZJ, Khaqan HA, Khauv P, Kheir WJ, Khetan V, Khodabande A, Khotenashvili Z, Kim JW, Kim JH, Kiratli H, Kivelä TT, Klett A, Komba Palet JEK, Krivaitiene D, Kruger M, Kulvichit K, Kuntorini MW, Kyara A, Lachmann ES, Lam CPS, Lam GC, Larson SA, Latinovic S, Laurenti KD, Le BHA, Lecuona K, Leverant AA, Li C, Limbu B, Long QB, López JP, Lukamba RM, Lumbroso L, Luna-Fineman S, Lutfi D, Lysytsia L, Magrath GN, Mahajan A, Majeed AR, Maka E, Makan M, Makimbetov EK, Manda C, Martín Begue N, Mason L, Mason JO, Matende IO, Materin M, Mattosinho CCDS, Matua M, Mayet I, Mbumba FB, McKenzie JD, Medina-Sanson A, Mehrvar A, Mengesha AA, Menon V, Mercado GJVD, Mets MB, Midena E, Mishra DKC, Mndeme FG, Mohamedani AA, Mohammad MT, Moll AC, Montero MM, Morales RA, Moreira C, Mruthyunjaya P, Msina MS, Msukwa G, Mudaliar SS, Muma KI, Munier FL, Murgoi G, Murray TG, Musa KO, Mushtaq A, Mustak H, Muyen OM, Naidu G, Nair AG, Naumenko L, Ndoye Roth PA, Nency YM, Neroev V, Ngo H, Nieves RM, Nikitovic M, Nkanga ED, Nkumbe H, Nuruddin M, Nyaywa M, Obono-Obiang G, Oguego NC, Olechowski A, Oliver SCN, Osei-Bonsu P, Ossandon D, Paez-Escamilla MA, Pagarra H, Painter SL, Paintsil V, Paiva L, Pal BP, Palanivelu MS, Papyan R, Parrozzani R, Parulekar M, Pascual Morales CR, Paton KE, Pawinska-Wasikowska K, Pe’er J, Peña A, Peric S, Pham CTM, Philbert R, Plager DA, Pochop P, Polania RA, Polyakov VG, Pompe MT, Pons JJ, Prat D, Prom V, Purwanto I, Qadir AO, Qayyum S, Qian J, Rahman A, Rahman S, Rahmat J, Rajkarnikar P, Ramanjulu R, Ramasubramanian A, Ramirez-Ortiz MA, Raobela L, Rashid R, Reddy MA, Reich E, Renner LA, Reynders D, Ribadu D, Riheia MM, Ritter-Sovinz P, Rojanaporn D, Romero L, Roy SR, Saab RH, Saakyan S, Sabhan AH, Sagoo MS, Said AMA, Saiju R, Salas B, San Román Pacheco S, Sánchez GL, Sayalith P, Scanlan TA, Schefler AC, Schoeman J, Sedaghat A, Seregard S, Seth R, Shah AS, Shakoor SA, Sharma MK, Sherief ST, Shetye NG, Shields CL, Siddiqui SN, Sidi Cheikh S, Silva S, Singh AD, Singh N, Singh U, Singha P, Sitorus RS, Skalet AH, Soebagjo HD, Sorochynska T, Ssali G, Stacey AW, Staffieri SE, Stahl ED, Stathopoulos C, Stirn Kranjc B, Stones DK, Strahlendorf C, Suarez MEC, Sultana S, Sun X, Sundy M, Superstein R, Supriyadi E, Surukrattanaskul S, Suzuki S, Svojgr K, Sylla F, Tamamyan G, Tan D, Tandili A, Tarrillo Leiva FF, Tashvighi M, Tateshi B, Tehuteru ES, Teixeira LF, Teh KH, Theophile T, Toledano H, Trang DL, Traoré F, Trichaiyaporn S, Tuncer S, Tyau-Tyau H, Umar AB, Unal E, Uner OE, Urbak SF, Ushakova TL, Usmanov RH, Valeina S, van Hoefen Wijsard M, Varadisai A, Vasquez L, Vaughan LO, Veleva-Krasteva NV, Verma N, Victor AA, Viksnins M, Villacís Chafla EG, Vishnevskia-Dai V, Vora T, Wachtel AE, Wackernagel W, Waddell K, Wade PD, Wali AH, Wang YZ, Weiss A, Wilson MW, Wime ADC, Wiwatwongwana A, Wiwatwongwana D, Wolley Dod C, Wongwai P, Xiang D, Xiao Y, Yam JC, Yang H, Yanga JM, Yaqub MA, Yarovaya VA, Yarovoy AA, Ye H, Yousef YA, Yuliawati P, Zapata López AM, Zein E, Zhang C, Zhang Y, Zhao J, Zheng X, Zhilyaeva K, Zia N, Ziko OAO, Zondervan M, Bowman R.. Global Retinoblastoma Presentation and Analysis by National Income Level. JAMA Oncol. 2020;6(5):685-695. doi:10.1001/jamaoncol.2019.6716. PMID:32105305; PMCID:PMC7047856.
متن مقاله را کپی کنید و در دستیار هوش مصنوعی دلخواه خود بچسبانید.
مقاله در کلیپبورد کپی شد
یکی از دستیارهای هوش مصنوعی زیر را باز کنید و متن کپیشده را در کادر گفتگو بچسبانید.