Retinoblastoma is a malignant tumor of the retina in infants and young children. It results from malignant transformation and proliferation of immature retinal cells, and is considered a monogenic disease caused by mutations in the RB1 gene located on the long arm of chromosome 13 (13q14.2). There is no sex predilection, and 95% are diagnosed by age 5.
The incidence is 1 in 15,000 to 23,000 live births, with 70-80 new cases per year in Japan. The ratio of unilateral to bilateral cases is 3:2, with bilateral cases diagnosed earlier (mean 8 months) than unilateral (mean 21 months). In developed countries, the 5-year survival rate for intraocular disease is over 95%.
Retinoblastoma is broadly classified into two types based on the type of genetic mutation.
Classification
Type of mutation
Characteristics of the disease
Genetic risk
Hereditary (germline mutation)
Germline RB1 mutation
Often bilateral and multifocal tumors
50% risk of inheritance in offspring
Non-hereditary (somatic mutation)
Somatic mutation in one retinal cell
Unilateral and solitary tumor
No inheritance to next generation
Hereditary (germline mutation): The first hit mutation is present in all cells of the body. Cancer develops when a second hit occurs. It tends to cause bilateral and multifocal tumors, and is inherited by 1 in 2 children (50%). There is a risk of secondary cancers such as osteosarcoma (15.7% at 20 years).
Non-hereditary (somatic mutation): When mutations occur in both alleles of the RB1 gene in a single retinal cell. It presents as a unilateral, solitary tumor, with no risk of inheritance to the next generation.
However, some unilateral cases include germline RB1 mutations. Even if the disease is unilateral, heritability should not be denied; family history, age at onset, and number of tumors must be interpreted based on genetic counseling and genetic evaluation. 1)
Staging directly determines the strategy for eye-preserving treatment.
Stage
Tumor Status
Estimated Eye Preservation Rate
T1 (Early intraocular)
Confined to eye, no progression
>90%
T2 (Advanced intraocular)
Intraocular progression
Approximately 50%
T3
Advanced with extraocular invasion
Approximately 10%
QIs retinoblastoma hereditary?
A
About 40% of cases are hereditary (germline RB1 mutation), with a 50% chance of passing it to offspring. The remaining 60% are non-hereditary (somatic mutation only) with no risk of inheritance. Hereditary cases tend to be bilateral and multifocal. Genetic testing and genetic counseling are recommended at diagnosis.
Aerts I, et al. Retinoblastoma. Orphanet J Rare Dis. 2006 Aug 25; 1:31. Figure 2. PMCID: PMC1586012. License: CC BY.
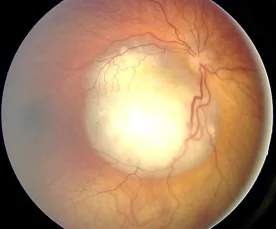
Fundus image of retinoblastoma observed as a white elevated lesion with rich vasculature, showing typical findings that cause leukocoria. Corresponds to the fundus findings and leukocoria discussed in section “2. Main Symptoms and Clinical Findings”.
In many cases, the tumor grows large within the eye and is discovered due to leukocoria. When it occurs in the macula, it may cause poor vision and strabismus, leading to detection. In older children, subjective vision loss may be the initial symptom, while in young children, rubbing the eye with poor vision may be the first sign.
Early Symptoms
Leukocoria: The most common initial symptom. The tumor enlarges inside the eye, causing the pupil to appear white.
Strabismus: Caused by poor vision due to a tumor in the macula. The eye with poor vision deviates outward.
Subjective vision loss: Seen in older children.
Eye rubbing: Seen in young children with poor vision.
Advanced Symptoms
Corneal opacity and increased intraocular pressure: Caused by lens displacement due to the tumor or increased intraocular pressure from neovascular glaucoma (NVG).
Conjunctival injection and eyelid swelling: Seen as inflammatory signs.
Pain: Occurs with elevated intraocular pressure or tumor necrosis.
Proptosis: Observed when extraocular extension occurs.
A vascular, white elevated lesion is observed; if calcification is present, diagnosis is straightforward. Vitreous seeding (tumor cells breaking off and spreading into the vitreous) is often present.
The red reflex test is fundamental for screening eye diseases in infants. The result is normal if both pupils are equal in size and show a bright, symmetric yellow-orange reflex. If the reflex is dark or too bright, or if there is asymmetry between the eyes, further evaluation is needed.
QDoes a white pupil always indicate retinoblastoma?
A
Causes of leukocoria (white pupil) include not only retinoblastoma but also persistent fetal vasculature (persistent hyperplastic primary vitreous), retinopathy of prematurity, Coats disease, and many others. However, if leukocoria is observed, prompt ophthalmologic evaluation to rule out retinoblastoma is the highest priority. Delay in diagnosis directly affects prognosis; therefore, if suspected, same-day referral is recommended.
Mutations in the RB1 gene on the long arm of chromosome 13 (13q14.2) are the cause. The RB1 gene produces the RB1 protein (retinoblastoma protein), which plays a key role in regulating cell division.
Each cell has two copies of the gene; a mutation in one copy alone does not impair function, but when both copies are mutated, the cell loses control of division and becomes malignant (two-hit hypothesis, Knudson hypothesis).
Hereditary (germline mutation): The first hit is a germline mutation (present in all cells). A second hit in a somatic cell leads to cancer. Bilateral and multifocal tumors are common.
Non-hereditary (somatic mutation): Both the first and second hits occur in a single somatic cell. This results in unilateral, unifocal tumors.
Family history is the greatest risk factor. The risk definitions recommended by the AAOOP (American Association of Ophthalmic Oncology and Pathology) are shown below 1).
Risk Category
Definition
Risk Value
High
Parent with bilateral Rb, or first- or second-degree relative with germline RB1 mutation carrier
In hereditary cases, attention must be paid to the risk of second cancers. Osteosarcoma is a typical example, occurring in 15.7% of hereditary cases within 20 years. Second cancers often develop after the teenage years.
Rumboldt Z, Dodig D, Galluzzi P, et al. Retinoblastoma and beyond: pediatric orbital mass lesions. Neuroradiology. 2025;67(2):469-492. Figure 2. PMID: 39729290; PMCID: PMC11893699; DOI: 10.1007/s00234-024-03517-6. License: CC BY.
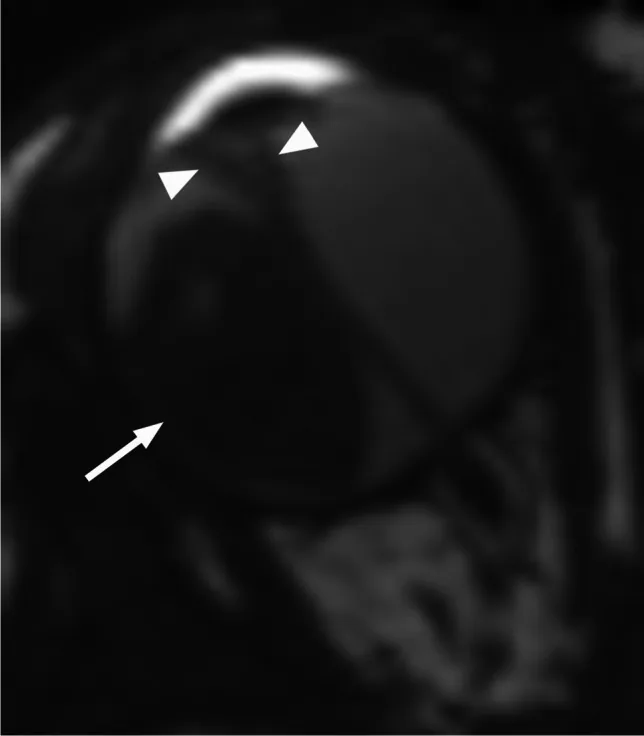
Axial T2-weighted MRI shows an exophytic retinoblastoma (arrow) with V-shaped secondary retinal detachment (arrowhead). This corresponds to the exophytic and endophytic growth patterns and MRI findings discussed in section “4. Diagnosis and Examination Methods.”
Biopsy of intraocular tumors is not performed. Intraocular lesions can be directly observed through transparent tissues, and clinical diagnosis is highly accurate. Additionally, biopsy of intraocular tumors can cause extraocular dissemination of tumor cells, leading to an unavoidable risk of metastasis. When eye-preserving treatment is planned, treatment is initiated based on clinical diagnosis.
Fundus examination (main): Clinical diagnosis confirmed by vascularized white elevated lesion with calcification
Ultrasonography: Confirmation of solid tumor and calcification. Note that calcification is less common in children over 5 years old.
MRI: Intermediate signal on T1, mildly low signal on T2, with contrast enhancement. Essential for evaluating optic nerve invasion and extraocular extension.
Head MRI: Screening is mandatory because about 3% of bilateral cases develop trilateral retinoblastoma (pineal gland tumor).
CT: Excellent for detecting calcification but involves radiation exposure. If MRI is available, CT is supplementary.
Coats disease: Yellow-white subretinal exudate; different tumor vascular pattern
Astrocytic hamartoma: Differentiated by presence of tumor vessels, OCT location, and growth
Intraocular cysticercosis: Rarely mimics Rb. A case report of a 4-year-old boy with leukocoria suspected as Rb underwent enucleation, and pathology revealed cysticercosis3)
For screening of children with familial Rb, the AAOOP 2018 recommendations are widely referenced internationally1).
Risk
Screening Schedule
End Time
High (>7.5%)
Birth to 8 weeks: every 2–4 weeks → 8–12 weeks: monthly → 1–2 years: every 2 months → 2–3 years: every 3 months → 3–4 years: every 4 months → 4–7 years: every 6 months
7 years (lifelong for RB1 mutation carriers)
Intermediate (1–7.5%)
Birth to 3 months: monthly → gradual reduction
7 years
Low (<1%)
Birth to 3 months: once a month → gradual reduction
7 years old
A retrospective cohort study using nationwide data from the Netherlands (1991–2019, 38 of 332 patients with familial Rb) showed that all 28 patients who received complete screening were diagnosed before 1 year of age (median 18 days), whereas the 10 patients with incomplete screening had a significantly delayed diagnosis with a median of 420 days (range 59 days to 4.8 years) 2). A protocol revision to reduce the screening end age to 2 years for low-risk groups (<3%) has also been proposed 2).
In familial cases, continuing fundus screening from immediately after birth directly affects prognosis. Classical registry studies also show that the timing of diagnosis in familial cases is closely related to screening frequency, and current practice is moving toward individualizing the end time by combining the presence or absence of RB1 mutations. 1, 2)
QIf there is a family history of retinoblastoma, until when is screening needed for the child?
A
The AAOOP recommends regular fundus examinations until age 7. With complete screening, most cases are diagnosed before 1 year of age. If genetic testing rules out RB1 mutation risk, early termination of screening is possible. For RB1 mutation carriers, irregular follow-up every 1–2 years after age 7 is recommended.
For early intraocular lesions where visual function can be expected, eye-preserving treatment is actively performed. In advanced intraocular stages, visual function is often not expected, but if the family wishes, eye-preserving treatment may be considered. Treatment requires a high level of expertise, and early referral to a specialized center is important.
Tumors up to about 3 mm in diameter are eligible. Direct irradiation with an infrared laser achieves local control in about 90% of cases. If the tumor is located in the macula, systemic chemotherapy is recommended first to avoid irreversible visual impairment.
② Cryotherapy
Tumors up to about 3 mm in the equatorial region or periphery are treated. The triple freeze-thaw method, repeating freezing and thawing three times, is common, and local control rates of about 90% are achieved, similar to laser therapy.
③ Brachytherapy
Indications: tumor thickness ≤5 mm, largest diameter ≤15 mm, localized tumor away from the optic disc. In Japan and Europe, ¹⁰⁶Ru (ruthenium-106, beta emitter) is used; in North America, ¹²⁵I seeds are used. The radioactive source is temporarily sutured onto the sclera overlying the tumor. This treatment requires a special treatment room and is available only at limited facilities. Local control rates of 80–90% can be achieved.
④ Systemic chemotherapy (VEC therapy)
First-line treatment for intraocular advanced-stage tumors. Triple-drug combination chemotherapy is widely used, but cure with chemotherapy alone is less than 10%. After tumor shrinkage, consolidation is performed with local treatments (laser, cryotherapy, brachytherapy).
Drug
Dose (body surface area based)
Dose (weight based for ≤36 months)
Schedule
Vincristine (Oncovin®)
1.5 mg/m²
0.05 mg/kg
day 1
Carboplatin (Paraplatin®)
560 mg/m²
18.6 mg/kg
day 1
Etoposide (Vepesid®)
150 mg/m²
5 mg/kg
day 1, 2
Repeat every 3–4 weeks for 2–6 cycles (all intravenous infusion).
A catheter is used to deliver the drug (melphalan; Alkeran® injection) directly into the ophthalmic artery. By delivering a higher dose locally to the eye and reducing systemic exposure, side effects such as bone marrow suppression can be reduced. Although not covered by insurance, it is an investigational treatment performed in more than 20 countries worldwide.
For vitreous seeding, systemic chemotherapy and intra-arterial infusion have limited efficacy, so intravitreal injection of melphalan (Alkeran® injection) is used in combination. This is an investigational treatment not covered by insurance.
⑦ External beam radiation therapy
Fractionated irradiation with 40–46 Gy of X-rays. Until the 1990s, this was the mainstay of eye-preserving treatment, but orbital bone deformity and increased risk of secondary cancers became evident. Currently, it is used only when other treatments cannot control the disease.
Positive optic nerve margin or extrascleral invasion are absolute indications for adjuvant therapy, and systemic chemotherapy and radiotherapy are performed. Marked choroidal invasion or optic nerve invasion beyond the lamina cribrosa are evaluated as relative risk factors for metastasis.
Anterior chamber or iris invasion: Risk of extraocular dissemination
When visual function recovery is not expected: Priority given to life prognosis
QIs it possible to preserve the eye?
A
For early intraocular tumors (T1), eye preservation is possible in over 90% of cases. Systemic chemotherapy (VEC regimen) is used to shrink the tumor, followed by consolidation with laser photocoagulation, cryotherapy, or brachytherapy. In advanced cases (T3), the eye preservation rate is about 10%, and enucleation may be necessary. The choice of treatment is determined by the specialist based on the stage and expected visual function.
The RB1 gene located on the long arm of chromosome 13 (13q14.2) encodes the RB1 protein (pRb), which plays a crucial role in regulating cell division. pRb binds to E2F transcription factors and suppresses the G1/S transition of the cell cycle, thereby controlling cell proliferation as a tumor suppressor protein.
According to the two-hit hypothesis proposed by Knudson, malignant transformation occurs when both alleles of the RB1 gene in a single cell are inactivated.
Hereditary: The first hit (germline mutation) is present in all cells. When a second hit (somatic mutation, LOH, etc.) occurs in a retinal cell, cancer develops. Therefore, bilateral and multifocal tumors are common, and diagnosis tends to occur relatively early.
Non-hereditary: Both the first and second hits occur as somatic mutations within the same retinal cell. Since the probability of both hits coinciding in one cell is low, tumors are usually unilateral and unifocal, and diagnosis tends to occur later than in hereditary cases.
In hereditary cases, the first hit of RB1 is present in all cells of the body. When a second hit occurs in cells outside the retina (e.g., bone, soft tissue), a second primary malignant tumor develops. Osteosarcoma is the most common, often occurring after the teenage years. In hereditary cases treated with external beam radiation therapy, the risk of second malignancies is further increased; therefore, external beam radiation is now used only in limited situations.
IAC (intra-arterial chemotherapy) delivers high-concentration drugs directly into the ophthalmic artery, minimizing systemic toxicity while achieving high intraocular drug concentration. It can expand the opportunity for eye preservation even in advanced cases with vitreous seeding. Large-scale cohort studies are ongoing to evaluate long-term outcomes.
Intravitreal melphalan injection is used for vitreous seeding, but international standardization of dosage and interval protocols remains a challenge. Multiple case series have reported high rates of seeding control, and future prospective trials are expected to establish its role.
A Dutch cohort study clearly demonstrated a correlation between complete screening attendance and early diagnosis (median 18 days), quantifying the adverse effects of screening interruption 2). Proposals to shorten the screening termination age (to 2 years) for low-risk groups are advancing protocol optimization based on risk stratification 2). Global dissemination of AAOOP recommendations and standardization of regional protocols remain future challenges 1).
Although rare, there are case reports of retinoblastoma coexisting with congenital brain malformations or chromosomal abnormalities. A report of bilateral retinoblastoma with Dandy-Walker syndrome highlights the importance of systemic evaluation including neurodevelopmental background, not just ocular symptoms. 4)
The widespread use of next-generation sequencing (NGS) has improved the detection sensitivity of germline RB1 mutations. The potential of monitoring using liquid biopsy (circulating tumor DNA) is also being studied, and its application to prognostic stratification without invasive biopsy is expected.
Standardization of second cancer surveillance protocols for long-term survivors of hereditary retinoblastoma is needed. In particular, the frequency and termination timing of multi-organ screening using whole-body MRI are being investigated through ongoing cohort studies to accumulate evidence.
Selective ophthalmic artery infusion (IAC) and intravitreal chemotherapy (IVitC) are becoming internationally established as treatments that improve eye preservation rates in advanced cases and those with vitreous seeding. In reviews of conservative treatment, they are positioned as strategies that expand eye preservation without increasing metastasis risk, and the implementation status and centralization of specialized facilities in each country affect outcomes. 5)
While the 5-year survival rate in developed countries is over 95%, reports indicate that in low- and middle-income countries in Africa and Asia, it remains at 25–70%. Systematic reviews and analyses by country income level consistently show disparities in survival and eye preservation rates, with late diagnosis, lack of access to healthcare, and shortage of specialized facilities cited as major factors. The dissemination of community-based screening programs, including the red reflex test, is an international challenge. 6, 7)
Skalet AH, Gombos DS, Gallie BL, Kim JW, Shields CL, Marr BP, Plon SE, Chévez-Barrios P. Screening Children at Risk for Retinoblastoma: Consensus Report from the American Association of Ophthalmic Oncologists and Pathologists. Ophthalmology. 2018;125(3):453-458. doi:10.1016/j.ophtha.2017.09.001. PMID:29056300.
Badalova NA, van Hoefen Wijsard M, Dommering CJ, et al. At what age could screening for familial retinoblastoma be stopped? Revised Dutch retrospective population-based cohort study 1991-2019. Ophthalmology. 2024;131(10):1189-1196.
Jakati S, Vempuluru VS, Kaliki S. Intraocular Cysticercosis Masquerading as Cavitary Retinoblastoma. Ophthalmology. 2025;132(4):e68. doi:10.1016/j.ophtha.2024.05.017. PMID:38878044.
Lomi N, Das D, Chawla B, Parampalli Ravindra A. Retinoblastoma in Dandy-Walker Syndrome. Cureus. 2025;17(8):e89663. doi:10.7759/cureus.89663. PMID:40926918; PMCID:PMC12415497.
Francis L. Munier, Maja Beck-Popovic, Guillermo L. Chantada, David Cobrinik, Tero T. Kivelä, Dietmar Lohmann, Philippe Maeder, Annette C. Moll, et al. Conservative management of retinoblastoma: Challenging orthodoxy without compromising the state of metastatic grace. “Alive, with good vision and no comorbidity”. Progress in Retinal and Eye Research. 2019;73:100764. doi:10.1016/j.preteyeres.2019.05.005.
Wong ES, Choy RW, Zhang Y, et al. Global retinoblastoma survival and globe preservation: a systematic review and meta-analysis. Lancet Glob Health. 2022;10(3):e380-e389.
Global Retinoblastoma Study Group, Fabian ID, Abdallah E, Abdullahi SU, Abdulqader RA, Adamou Boubacar S, Ademola-Popoola DS, Adio A, Afshar AR, Aggarwal P, Aghaji AE, Ahmad A, Akib MNR, Al Harby L, Al Ani MH, Alakbarova A, Portabella SA, Al-Badri SAF, Alcasabas APA, Al-Dahmash SA, Alejos A, Alemany-Rubio E, Alfa Bio AI, Alfonso Carreras Y, Al-Haddad C, Al-Hussaini HHY, Ali AM, Alia DB, Al-Jadiry MF, Al-Jumaily U, Alkatan HM, All-Eriksson C, Al-Mafrachi AARM, Almeida AA, Alsawidi KM, Al-Shaheen AASM, Al-Shammary EH, Amiruddin PO, Antonino R, Astbury NJ, Atalay HT, Atchaneeyasakul LO, Atsiaya R, Attaseth T, Aung TH, Ayala S, Baizakova B, Balaguer J, Balayeva R, Balwierz W, Barranco H, Bascaran C, Beck Popovic M, Benavides R, Benmiloud S, Bennani Guebessi N, Berete RC, Berry JL, Bhaduri A, Bhat S, Biddulph SJ, Biewald EM, Bobrova N, Boehme M, Boldt HC, Bonanomi MTBC, Bornfeld N, Bouda GC, Bouguila H, Boumedane A, Brennan RC, Brichard BG, Buaboonnam J, Calderón-Sotelo P, Calle Jara DA, Camuglia JE, Cano MR, Capra M, Cassoux N, Castela G, Castillo L, Català-Mora J, Chantada GL, Chaudhry S, Chaugule SS, Chauhan A, Chawla B, Chernodrinska VS, Chiwanga FS, Chuluunbat T, Cieslik K, Cockcroft RL, Comsa C, Correa ZM, Correa Llano MG, Corson TW, Cowan-Lyn KE, Csóka M, Cui X, Da Gama IV, Dangboon W, Das A, Das S, Davanzo JM, Davidson A, De Potter P, Delgado KQ, Demirci H, Desjardins L, Diaz Coronado RY, Dimaras H, Dodgshun AJ, Donaldson C, Donato Macedo CR, Dragomir MD, Du Y, Du Bruyn M, Edison KS, Eka Sutyawan IW, El Kettani A, Elbahi AM, Elder JE, Elgalaly D, Elhaddad AM, Elhassan MMA, Elzembely MM, Essuman VA, Evina TGA, Fadoo Z, Fandiño AC, Faranoush M, Fasina O, Fernández DDPG, Fernández-Teijeiro A, Foster A, Frenkel S, Fu LD, Fuentes-Alabi SL, Gallie BL, Gandiwa M, Garcia JL, García Aldana D, Gassant PY, Geel JA, Ghassemi F, Girón AV, Gizachew Z, Goenz MA, Gold AS, Goldberg-Lavid M, Gole GA, Gomel N, Gonzalez E, Gonzalez Perez G, González-Rodríguez L, Garcia Pacheco HN, Graells J, Green L, Gregersen PA, Grigorovski NDAK, Guedenon KM, Gunasekera DS, Gündüz AK, Gupta H, Gupta S, Hadjistilianou T, Hamel P, Hamid SA, Hamzah N, Hansen ED, Harbour JW, Hartnett ME, Hasanreisoglu M, Hassan S, Hassan S, Hederova S, Hernandez J, Hernandez LMC, Hessissen L, Hordofa DF, Huang LC, Hubbard GB, Hummlen M, Husakova K, Hussein Al-Janabi AN, Ida R, Ilic VR, Jairaj V, Jeeva I, Jenkinson H, Ji X, Jo DH, Johnson KP, Johnson WJ, Jones MM, Kabesha TBA, Kabore RL, Kaliki S, Kalinaki A, Kantar M, Kao LY, Kardava T, Kebudi R, Kepak T, Keren-Froim N, Khan ZJ, Khaqan HA, Khauv P, Kheir WJ, Khetan V, Khodabande A, Khotenashvili Z, Kim JW, Kim JH, Kiratli H, Kivelä TT, Klett A, Komba Palet JEK, Krivaitiene D, Kruger M, Kulvichit K, Kuntorini MW, Kyara A, Lachmann ES, Lam CPS, Lam GC, Larson SA, Latinovic S, Laurenti KD, Le BHA, Lecuona K, Leverant AA, Li C, Limbu B, Long QB, López JP, Lukamba RM, Lumbroso L, Luna-Fineman S, Lutfi D, Lysytsia L, Magrath GN, Mahajan A, Majeed AR, Maka E, Makan M, Makimbetov EK, Manda C, Martín Begue N, Mason L, Mason JO, Matende IO, Materin M, Mattosinho CCDS, Matua M, Mayet I, Mbumba FB, McKenzie JD, Medina-Sanson A, Mehrvar A, Mengesha AA, Menon V, Mercado GJVD, Mets MB, Midena E, Mishra DKC, Mndeme FG, Mohamedani AA, Mohammad MT, Moll AC, Montero MM, Morales RA, Moreira C, Mruthyunjaya P, Msina MS, Msukwa G, Mudaliar SS, Muma KI, Munier FL, Murgoi G, Murray TG, Musa KO, Mushtaq A, Mustak H, Muyen OM, Naidu G, Nair AG, Naumenko L, Ndoye Roth PA, Nency YM, Neroev V, Ngo H, Nieves RM, Nikitovic M, Nkanga ED, Nkumbe H, Nuruddin M, Nyaywa M, Obono-Obiang G, Oguego NC, Olechowski A, Oliver SCN, Osei-Bonsu P, Ossandon D, Paez-Escamilla MA, Pagarra H, Painter SL, Paintsil V, Paiva L, Pal BP, Palanivelu MS, Papyan R, Parrozzani R, Parulekar M, Pascual Morales CR, Paton KE, Pawinska-Wasikowska K, Pe’er J, Peña A, Peric S, Pham CTM, Philbert R, Plager DA, Pochop P, Polania RA, Polyakov VG, Pompe MT, Pons JJ, Prat D, Prom V, Purwanto I, Qadir AO, Qayyum S, Qian J, Rahman A, Rahman S, Rahmat J, Rajkarnikar P, Ramanjulu R, Ramasubramanian A, Ramirez-Ortiz MA, Raobela L, Rashid R, Reddy MA, Reich E, Renner LA, Reynders D, Ribadu D, Riheia MM, Ritter-Sovinz P, Rojanaporn D, Romero L, Roy SR, Saab RH, Saakyan S, Sabhan AH, Sagoo MS, Said AMA, Saiju R, Salas B, San Román Pacheco S, Sánchez GL, Sayalith P, Scanlan TA, Schefler AC, Schoeman J, Sedaghat A, Seregard S, Seth R, Shah AS, Shakoor SA, Sharma MK, Sherief ST, Shetye NG, Shields CL, Siddiqui SN, Sidi Cheikh S, Silva S, Singh AD, Singh N, Singh U, Singha P, Sitorus RS, Skalet AH, Soebagjo HD, Sorochynska T, Ssali G, Stacey AW, Staffieri SE, Stahl ED, Stathopoulos C, Stirn Kranjc B, Stones DK, Strahlendorf C, Suarez MEC, Sultana S, Sun X, Sundy M, Superstein R, Supriyadi E, Surukrattanaskul S, Suzuki S, Svojgr K, Sylla F, Tamamyan G, Tan D, Tandili A, Tarrillo Leiva FF, Tashvighi M, Tateshi B, Tehuteru ES, Teixeira LF, Teh KH, Theophile T, Toledano H, Trang DL, Traoré F, Trichaiyaporn S, Tuncer S, Tyau-Tyau H, Umar AB, Unal E, Uner OE, Urbak SF, Ushakova TL, Usmanov RH, Valeina S, van Hoefen Wijsard M, Varadisai A, Vasquez L, Vaughan LO, Veleva-Krasteva NV, Verma N, Victor AA, Viksnins M, Villacís Chafla EG, Vishnevskia-Dai V, Vora T, Wachtel AE, Wackernagel W, Waddell K, Wade PD, Wali AH, Wang YZ, Weiss A, Wilson MW, Wime ADC, Wiwatwongwana A, Wiwatwongwana D, Wolley Dod C, Wongwai P, Xiang D, Xiao Y, Yam JC, Yang H, Yanga JM, Yaqub MA, Yarovaya VA, Yarovoy AA, Ye H, Yousef YA, Yuliawati P, Zapata López AM, Zein E, Zhang C, Zhang Y, Zhao J, Zheng X, Zhilyaeva K, Zia N, Ziko OAO, Zondervan M, Bowman R.. Global Retinoblastoma Presentation and Analysis by National Income Level. JAMA Oncol. 2020;6(5):685-695. doi:10.1001/jamaoncol.2019.6716. PMID:32105305; PMCID:PMC7047856.
Copy the article text and paste it into your preferred AI assistant.
Article copied to clipboard
Open an AI assistant below and paste the copied text into the chat box.