Stage 1–2
Stage 1: Only retinal telangiectasia is present, without exudation.
Stage 2A: In addition to telangiectasia, exudates are present outside the fovea.
Stage 2B: Exudates extend to the fovea. Visual acuity decline becomes apparent.

Coats disease is an idiopathic retinal vascular disease reported by George Coats in 1908. Its essence is abnormal dilation of retinal capillaries (telangiectasia) and accumulation of exudates within and beneath the retina from the vessel walls.
The disease is sporadic and non-hereditary, with no association with systemic diseases or family history 1). The incidence is rare at 0.09 per 100,000 people 2). Approximately 75% of patients are male, 95% have unilateral disease, and it commonly occurs in individuals under 20 years (average around 5 years).
Adult onset is very rare but presents a different clinical picture from the childhood type. Adult-onset cases are milder and slower in progression than the childhood type, and treatment response is also favorable 3). In one report, only 21% of 48 adult-onset eyes developed exudative retinal detachment, significantly lower than 81% in childhood-onset cases 3).
The mild form is also called Leber’s miliary aneurysms and forms part of the spectrum of Coats disease. Type 1 idiopathic macular telangiectasia is also considered to be within the same disease spectrum.
Cases of onset at age 35 or older have been reported and are recognized as adult-onset Coats disease 3). Compared to the childhood type, the progression of lesions is slower, the frequency of exudative retinal detachment is lower, and treatment response is often favorable. However, onset itself is rare, and when similar findings are observed in adults, differentiation from other diseases is important.
When onset occurs in infancy, the affected child rarely complains of symptoms.
In adult-onset cases, vision is relatively preserved, and it may be discovered incidentally during health checkups 3).
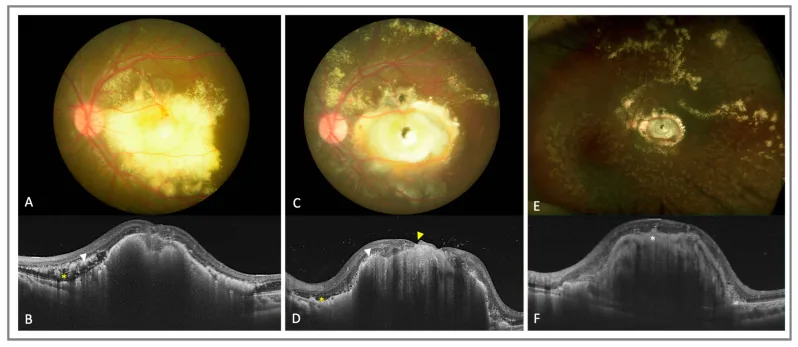
Characteristic fundus findings include abnormally dilated retinal vessels concentrated in the periphery and accumulation of yellowish-white subretinal and intraretinal exudates. Fluorescein angiography reveals peripheral retinal vascular occlusion, telangiectasia, microaneurysms, and neovascularization as characteristic findings. As the disease progresses, exudative retinal detachment occurs, eventually leading to total detachment. On ultrasound, CT, and MRI, subretinal fluid appears uniformly throughout, which is an important imaging differential point from retinoblastoma, which presents with a heterogeneous appearance with calcification.
In later stages, anterior segment complications such as rubeosis iridis and neovascular glaucoma occur. Macular hole is a rare complication, with only about 7 cases reported in the literature 4). Fibrous nodules are a factor for poor visual prognosis, and OCTA has confirmed type 3 neovascularization (SVC→DVC→avascular complex) within the nodules 5).
The Shields classification divides the stages of Coats disease into the following 5 stages. It is used for selecting treatment strategies and estimating prognosis.
Stage 1–2
Stage 1: Only retinal telangiectasia is present, without exudation.
Stage 2A: In addition to telangiectasia, exudates are present outside the fovea.
Stage 2B: Exudates extend to the fovea. Visual acuity decline becomes apparent.
Stage 3
Stage 3A1: Associated with subtotal retinal detachment outside the fovea.
Stage 3A2: Associated with subtotal retinal detachment involving the fovea. Visual prognosis is poor.
Stage 3B: Total retinal detachment has occurred. Urgent intervention is required.
Stages 4–5
Stage 4: Total retinal detachment complicated by secondary glaucoma, a pre-end-stage condition. May be painful.
Stage 5: End stage. Phthisis bulbi (atrophy) has occurred. Enucleation may be considered.
Several diseases present with a white pupil, and Coats disease is only one of them. The most important differential diagnosis is retinoblastoma (Rb), which requires urgent evaluation as it directly affects life prognosis. Other differentials include retinopathy of prematurity, persistent hyperplastic primary vitreous (PHPV), and endophthalmitis. For details, see the “Diagnosis and Testing Methods” section.
The cause of Coats disease is unknown, and no association with systemic diseases or family history has been identified. Reports suggesting a genetic background have indicated instability of chromosomes 3 and 13, and associations with the NDP gene (Norrie disease-related) and CRB1 gene, but these are not established.
As the starting point of the pathology, disruption of the inner blood-retinal barrier (iBRB) is thought to play a central role1). Decreased pericytes (cells that support vascular endothelium) weaken the vessel walls, leading to the formation of abnormally dilated capillaries and aneurysms1). Plasma components leak and accumulate within the vessel wall and retinal layers, causing thickening of the vessel wall and further exudation, creating a vicious cycle2).
A high VEGF environment is also suggested to promote dilation of peripheral capillaries2), and this mechanism provides the theoretical basis for anti-VEGF therapy.
Diagnosis involves a combination of multiple tests, and the most important task is to reliably differentiate it from retinoblastoma (Rb).
Fundus examination under mydriasis reveals tortuous abnormal vascular networks and yellowish-white subretinal exudates in the peripheral retina. When exudates extend to the macula, they may appear as hard white plaques.
This is one of the most important tests for diagnosing Coats disease. Characteristic findings include marked dilation of capillaries, capillary aneurysms, arteriovenous anastomoses, and “light bulbs” (bulbous fluorescein leakage). It is essential for determining the extent of abnormal vessels and planning laser photocoagulation.
Confirms the absence of a solid mass. Rb often shows hyperechoic foci (calcification) within a solid mass on B-scan, whereas Coats disease does not form a solid mass.
Evaluate the presence of calcification. Rb is frequently associated with calcification, while Coats disease does not cause calcification. This finding is an important basis for differentiation.
The main differences between Coats disease and retinoblastoma are shown below.
| Feature | Coats disease | Retinoblastoma |
|---|---|---|
| Typical age of onset | Around 5 years old | 1 to 2 years old |
| Sex predilection | 75% male | None |
| Bilaterality | About 5% | About 40% |
| Calcification | Absent | Present (high rate) |
| Solid mass | None | Present |
| Ultrasound | Subretinal fluid accumulation | Solid mass, internal calcification, posterior shadowing |
| MRI | Subretinal fluid is homogeneous | Heterogeneous (mass signal) |
Other differential diagnoses include retinal hemangioma, von Hippel-Lindau disease, persistent hyperplastic primary vitreous (PHPV), familial exudative vitreoretinopathy (FEVR), toxocariasis, vasoproliferative tumor, and Eales disease.
The goal of treatment is to occlude abnormal blood vessels and stop the production of exudates. A stepwise approach is taken depending on the disease stage.
This is the first-line treatment. Abnormal dilated vessels and microaneurysms identified by FA are directly coagulated, and photocoagulation is also applied to the surrounding non-perfusion areas. In children, it is performed under general anesthesia. Multiple sessions are often required, and regular FA re-evaluation and additional coagulation are repeated after treatment.
This is the next option for lesions in the anterior periphery or areas difficult to treat with photocoagulation. It may be used in combination with photocoagulation.
Laser Coagulation
Indications: Abnormal vessels and exudative lesions in Stages 1 to 3A.
Method: Direct coagulation of telangiectatic areas and non-perfusion regions under FA guidance. Performed under general anesthesia in children.
Features: Can be performed repeatedly. Regular FA re-evaluation and additional coagulation after treatment are standard management.
Cryocoagulation
Indications: Anterior peripheral lesions difficult to treat with photocoagulation, and as an adjunct in severe cases up to Stage 3B.
Method: A cryoprobe is applied transsclerally to coagulate and occlude abnormal vessels.
Features: It has the advantage of being feasible even under opaque media or in the far periphery.
Vitreoretinal Surgery
Indications: Stage 3B (total retinal detachment) or higher, and cases where cryocoagulation is ineffective.
Method: Drainage of subretinal fluid via external drainage or vitrectomy, and internal retinal reattachment 8). In severe cases, subretinal fluid drainage combined with scleral buckling may be performed.
Features: In cases with macular hole, the inverted internal limiting membrane (ILM) flap technique has been reported to be effective 4).
The use of anti-VEGF agents for Coats disease has not yet reached a consensus; it is not a standard treatment but is positioned as an adjuvant therapy in combination with photocoagulation.
A report describes a case of adult-onset Coats disease treated with a combination of intravitreal injection of ranibizumab 0.5 mg and laser photocoagulation, with final best-corrected visual acuity improving from counting fingers to 20/603).
In a pediatric case, combination therapy with intravitreal bevacizumab 1.25 mg, sub-Tenon triamcinolone, and laser was performed every 6 weeks under EUA (examination under anesthesia). It has been noted that paradoxical exudative retinopathy may occur after treatment2).
There is also a report that brolucizumab was effective in a case resistant to bevacizumab2). Anti-VEGF therapy may contribute to preventing the formation of fibrous nodules5).
Enucleation is chosen for painful blind eyes (Stage 4–5) when it is difficult to rule out retinoblastoma.
In Coats disease, some cases experience recurrence or re-detachment over several years, and some cases are bilateral with different onset times. Even after treatment completion, regular re-evaluation with fluorescein angiography is necessary, and if new lesions are found, additional photocoagulation or cryotherapy should be performed.
At present, there is no established consensus on the use of anti-VEGF drugs for Coats disease, and they are not considered standard treatment. Although reports are accumulating on their use as adjunctive therapy combined with laser photocoagulation, evaluation of efficacy and safety requires further research.
The central mechanism of Coats disease is the breakdown of the inner blood-retinal barrier (iBRB)1).
The iBRB is composed of retinal capillary endothelial cells and supporting pericytes. In Coats disease, the number of pericytes is markedly reduced, leading to decreased vascular endothelial support function1). Immunostaining and electron microscopy have confirmed that the number of endothelial cells themselves is also reduced1).
Breakdown of the endothelial BRB causes plasma components (mainly lipoproteins and cholesterol) to leak and accumulate within the vessel wall, retina, and subretinal space1). The accumulated lipids trigger infiltration of lipid-laden macrophages (foam cells) and a granulomatous immune response, exacerbating tissue damage1).
A high VEGF environment promotes further dilation of peripheral capillaries and contributes to disease progression2). OCTA observations have confirmed type 3 neovascularization (formed in the order SVC → DVC → avascular complex) within macular fibrous nodules in advanced lesions5), advancing the understanding of the neovascularization process.
The mechanism of macular hole formation is thought to involve retinal shortening due to peripheral laser photocoagulation, which creates tangential traction and leads to a macular perforation4).
OCTA has enabled non-invasive evaluation of microvascular structures within fibrous nodules.
Ong et al. (2021) used OCTA to analyze the vascular structure within macular nodules in detail, revealing the presence of type 3 neovascularization formed in the order SVC → DVC → avascular complex5). This finding is important for elucidating the mechanism of nodule formation and as a target for anti-VEGF therapy.
A phenomenon of paradoxical worsening of exudation after initiation of anti-VEGF therapy has been reported, and elucidation of the mechanism and establishment of management methods are needed.
Kalavar et al. (2022) reported a case of pediatric Coats disease with transient worsening of exudation and macular star formation after treatment initiation 2). Brolucizumab was effective in a bevacizumab-resistant case, attracting attention as a new treatment option 2).
Nawrocka et al. (2023) reported a case of vitrectomy using the inverted internal limiting membrane flap technique for a macular hole associated with Coats disease 4). Macular hole closure was confirmed at 18 months postoperatively, and final best-corrected visual acuity was 20/40. Coats disease-related macular hole is a rare complication with only about 7 cases reported on PubMed 4).
Shields et al. (2019) analyzed 351 eyes with Coats disease over 45 years, showing improvement in treatment outcomes over time 9). Dalvin et al. (2019) analyzed the same cohort by age category, showing that pediatric-onset cases tend to have more severe lesions and worse visual prognosis compared to adult-onset cases 10).
Adult-onset Coats disease is a disease concept that has been underrecognized, and the number of reports is increasing 3). Clinical differences from the pediatric type (milder, slowly progressive, good treatment response) are being clarified, and establishing appropriate treatment protocols for adults remains a challenge.